CuO/ZrO2催化水煤气变换反应制氢:ZrO2载体焙烧温度的影响
2019-04-18张燕杰陈崇启詹瑛瑛叶远松娄本勇郑国才
张燕杰, 陈崇启, 詹瑛瑛, 叶远松, 娄本勇, 郑国才, 林 棋
(1. 闽江学院 海洋学院, 福建 福州 350108; 2. 福州大学 化肥催化剂国家工程研究中心, 福建 福州 350002)
水煤气变换反应 [CO(g) + H2O(g)CO2(g) + H2(g),ΔH(298 K) = -41.2 kJ/mol,WGSR] 在消除CO的同时还可获得H2,主要用于工业制氢、合成氨及合成甲醇等传统化工领域。近年来,由于在质子交换膜燃料电池(PEMFCs)在线制氢系统中的重要作用,WGS反应再次引起广泛关注[1-3]。传统WGS催化剂包括Fe-Cr系高温变换催化剂、Cu-Zn-Al系低温变换催化剂和Co-Mo系宽温耐硫变换催化剂。但它们通常需要复杂的活化过程,易自燃,抗冷凝水及抗循环使用性能差,已无法满足PEMFCs的实际需求[4]。因此,迫切需要开发新型的WGS催化剂。
负载型金属催化剂由于可以获得较高的金属分散度和特殊的金属-载体协同作用,一直是WGS催化剂领域的研究重点[4]。对CO分子具有中等吸附能力的贵金属(如Au[5]、Pt[6,7]、Rh[8]、Ru[9]、Ir[10]等)和非贵金属(如Ni[11]和Cu[12-15])常被选作新型WGS催化剂的活性金属;而具有还原性能或表面容易产生氧空位的金属氧化物,如TiO2[7,11]、Fe2O3[10,13]、CeO2[14,15]、CeZrO4[3,6]、ZnO[16]和ZrO2[17,18]等,因对H2O分子的解离具有促进作用,常被选作催化剂载体。这其中,Cu对WGS反应的催化选择性和活性较高,且成本低廉,因此,受到较多关注[12-15]。同时,ZrO2具有特殊的物理化学性能(同时具有氧化性、还原性、表面酸性以及表面碱性)和优良的结构稳定性[19,20]。p型半导体的属性使ZrO2表面容易形成氧空位,这使得CuO-ZrO2之间较易产生较强的相互作用[21]。因此,CuO/ZrO2组合应是一种极具开发潜力的WGS催化剂。
Ko等[22]首次报道了CuO/ZrO2体系在催化WGS反应中应用,发现反应温度低于200 ℃时该体系表现出较传统Cu-Zn-Al催化剂更高的催化活性。CuO的分散状态[23,24]以及CuO-ZrO2之间的相互作用[23,25]被认为是影响CuO/ZrO2催化剂WGS催化性能的关键因素。由于ZrO2载体的结构和表面性质会对上述因素产生直接影响。因此,通过改变制备方法[23,24]、掺杂过渡金属离子[26]以及改变预处理方法[27,28]等手段预先调控ZrO2载体的结构和表面性质,成为制备高性能CuO/ZrO2及其他ZrO2基WGS催化剂的重要研究方法。此外,明确CuO/ZrO2催化剂的活性位结构对于指导催化剂的改进合成同样具有重要意义。近期,本课题组通过CO-TPR质谱跟踪表征结合催化反应动力学实验证实,与ZrO2表面具有强相互作用的高分散CuO物种(即Cu-[O]-Zr物种,“[ ]”代表ZrO2表面氧空位)是CuO/ZrO2催化剂上催化WGS反应的活性CuO物种,而与ZrO2表面不具有强相互作用的CuO簇和晶相CuO则并未参与催化WGS反应[29]。Cu-[O]-Zr物种的存在显著提高了ZrO2活性表面羟基与CO发生反应(即表面WGS反应)的能力,这是CuO/ZrO2具有优良WGS催化活性的主要原因[26,29]。
基于上述分析,本研究通过改变ZrO2的焙烧温度来调变ZrO2的结构和表面性质,并探究ZrO2的结构和表面性质对CuO/ZrO2催化剂上CuO的分散状态,CuO-ZrO2之间的相互作用及其WGS催化活性的影响,以深入揭示CuO/ZrO2WGS催化剂的构效关系。
1 实验部分
1.1 试剂
硝酸铜[Cu(NO3)2·3H2O]、氧氯化锆(ZrOCl2·8H2O)、氢氧化钾(KOH)及尿素[CO(NH2)2]均购于国药集团化学试剂有限公司。溶液采用去离子水配制。
1.2 实验过程
1.2.1催化剂的制备
ZrO2载体采用水热法制备。称取6.240 g尿素和16.757 g ZrOCl2·8H2O溶于少量水中并标定至60 mL,然后转入内衬为100 mL的水热反应釜中,再于150 ℃加热24 h。待反应釜降至室温后,将水热产物取出并离心洗涤数次,直至检测无Cl-残留。所得产物在120 ℃下干燥8 h,再分别置于马弗炉中于120、250、350和450 ℃焙烧4 h制得ZrO2载体,分别标记为Z-120、Z-250、Z-350及Z-450。
CuO/ZrO2催化剂采用沉积-沉淀法制备,催化剂中Cu的理论质量分数为8.2%。将1.012 g Cu(NO3)2·3H2O连同3 g研磨均匀的ZrO2载体一同分散于200 mL去离子水中。控制水浴温度为60 ℃,机械搅拌下向上述混合溶液中滴加0.5 mol/L的KOH水溶液至体系终点pH值为9.0。然后保持水浴温度60 ℃,继续搅拌1 h。所得沉淀物经多次离心洗涤除去杂质离子后再于120 ℃下干燥8 h,而后置于马弗炉中于400 ℃焙烧4 h得到CuO/ZrO2催化剂。根据ZrO2的焙烧温度,样品分别标记为C/Z-120、C/Z-250、C/Z-350及C/Z-450。
1.2.2催化剂的表征
使用X’Pert Pro粉末衍射仪(荷兰Panalytical公司)对样品的晶相结构进行分析。使用CoKα射线,λ为0.1790 nm,管电流为40 mA,管电压为40 kV,扫描步长为0.02°。
使用ASAP2020物理吸附仪(美国Micrometrics公司)对样品的比表面积和孔结构进行测试。液氮温度下(-196 ℃),以氮气为吸附质测定N2-物理吸附-脱附等温线。采用多点BET法和BJH法(取脱附支数据)分别计算样品的比表面积和孔结构。
H2程序升温还原(H2-TPR)实验在AutoChem 2910(美国Micrometric公司)化学吸附仪上进行。将50 mg样品于高纯He(30 mL/min)下200 ℃预处理30 min。降至室温后,气路切换为10.0% H2/Ar(30 mL/min)。待TCD信号基线平稳后,开始对样品进行程序升温还原,升温速率为10 ℃/min,测试温区为50-300 ℃。还原峰所对应的耗氢量通过对比H2和Ar混合气的TCD信号标准曲线求得。
H2O程序升温脱附及质谱跟踪(H2O-TPD-MS)实验在AutoChem II 2920化学吸附仪(美国Micrometric公司)上进行。准确称量50 mg样品,不经过高温预处理,在高纯He(30 mL/min)下,由室温开始对样品进行程序升温加热至400 ℃,升温速率为10 ℃/min。使用HPR20质谱仪(英国HIEDN公司)对尾气中的H2O(气体)(m/z= 18)进行跟踪,H2O(气体)经过的管线加装有热保护,以防止H2O(气体)冷凝。
采用N2O滴定法测定样品的Cu分散度和Cu金属比表面积[29],仪器使用AutoChem 2910 (美国Micrometric公司)化学吸附仪。将50 mg样品于高纯He(30 mL/min)下200 ℃预处理30 min。降至室温后,气路切换为10.0% H2/Ar(30 mL/min),对样品进行程序升温还原,升温速率10 ℃/min,终点温度为300 ℃,此时样品中的全部CuO均被还原为Cu单质[29]。而后在高纯He保护下,将样品冷却至60 ℃,吹扫30 min 3.35% N2O/He混合气,此时仅Cu单质的表面Cu原子发生如式(1)所示的反应,形成Cu2O,式中字母S指代表面原子。再次进行H2程序升温还原操作,终点温度同为300 ℃,此时发生式(2)所示反应,即Cu2O再次被还原成单质Cu。依据式(2)通过计算耗H2量即可确定表面Cu原子数量。样品的Cu分散度dCu采用式(3)计算,式中,n(S)代表表面Cu原子数,n(total)代表Cu原子总数(依据ICP-OES结果测算)。Cu金属比表面积ACu采用式(4)计算,式中mcat指代催化剂的质量,1.47×1019为每平方米上Cu原子的数目。
2Cu(S)+ N2O → Cu2O(S)+ N2
(1)
Cu2O(S)+ H2→ 2Cu(S)+ H2O
(2)
dCu=n(S)/n(total)× 100%
(3)
ACu=n(S)/(mcat× 1.47 × 1019)
(4)
CO程序升温还原及质谱跟踪(CO-TPR-MS)测试在AutoChem II 2920化学吸附仪(美国Micrometric公司)上进行。将50 mg样品于高纯He(30 mL/min)下200 ℃吹扫30 min。待降至室温后,气路切换为5.0% CO/He(40 mL/min),对样品进行程序升温还原至400 ℃,升温速率为10 ℃/min。使用HPR20质谱仪(英国HIEDN公司)对尾气中的CO(m/z= 18)、H2(m/z= 2)和CO2(m/z= 44)进行跟踪。
1.2.3催化剂的活性评价
使用CO-CMAT9001型固定床反应器对样品的活性进行评价。测试条件如下:常压,样品的装填量0.5 g,汽气体积比0.4∶1,质量空速4000 cm3/(g·h),测试温区为180-300 ℃,间隔为30 ℃。原料气(干气)组成为15% CO、55% H2、23% N2、7% CO2(体积分数)。反应尾气中CO的体积分数使用GC-8A型气相色谱仪(日本Shimadzu公司)在线分析。样品的WGS催化活性以CO转化率x表示:
(5)
式中,φ′为尾气中CO的体积分数,φ为原料气中CO的体积分数。
2 结果与讨论
2.1 ZrO2载体及CuO/ZrO2催化剂的结构表征
图1 (a)、图1 (b)为经不同温度焙烧的ZrO2载体及相应CuO/ZrO2催化剂的XRD谱图。图1 (a)中,各ZrO2载体的XRD谱线中均只出现单斜相ZrO2(即m-ZrO2)的特征衍射峰(JCPDS: 01-089-9066,标记为“◆”),表明在所研究的焙烧温度范围内ZrO2未发生相变。随着焙烧温度的升高,ZrO2的衍射峰强度逐渐增强、半高宽逐渐变窄,表明ZrO2的晶粒粒径逐渐增大(见表1)。其中,样品Z-450的晶粒粒径相较于Z-350的增大幅度尤为明显,表明在450 ℃下焙烧会导致ZrO2发生一定程度的烧结。由图1 (b)可知,系列CuO/ZrO2催化剂的XRD谱线中同时出现了m-ZrO2和单斜相CuO (JCPDS: 05-0661,标记为“▽”)的特征衍射峰。催化剂中ZrO2的晶粒粒径与ZrO2载体保持了相同的变化规律(见表1)。值得注意的是,CuO/ZrO2催化剂虽然于400 ℃经历了4 h的二次焙烧,其ZrO2晶粒的粒径与ZrO2载体相比却未明显增大。与样品Z-120和Z-250相比,样品C/Z-120和C/Z-250的ZrO2晶粒粒径甚至有所减小,这说明在催化剂焙烧过程中,负载CuO与ZrO2之间产生了一定的相互作用,这种作用抑制了ZrO2晶粒受热长大,且载体焙烧温度较低时,这种相互作用会更为显著。

图 1 经不同温度焙烧的ZrO2载体(a)及相应CuO/ZrO2催化剂(b)的XRD谱图
此外,由图1 (b)还可知,CuO的衍射峰强度随着ZrO2焙烧温度的升高逐渐增强,表明催化剂中晶相CuO的含量逐渐增多。如前言部分所述,采用与本研究相同方法制得的CuO/ZrO2催化剂,其表面存在三种形态的CuO物种:(α)高分散CuO簇;(β)与ZrO2表面紧密作用的高分散Cu-[O]-Zr物种;(γ)晶相CuO。鉴于本研究中系列催化剂的CuO含量基本相同(见表1),由此,根据不同样品晶相CuO含量的变化规律,可以推测催化剂中高分散型态CuO的含量(即α和β物种的总含量)应随着ZrO2焙烧温度的升高而逐渐减少,这将在后续H2-TPR和CO-TPR-MS部分进行进一步说明。
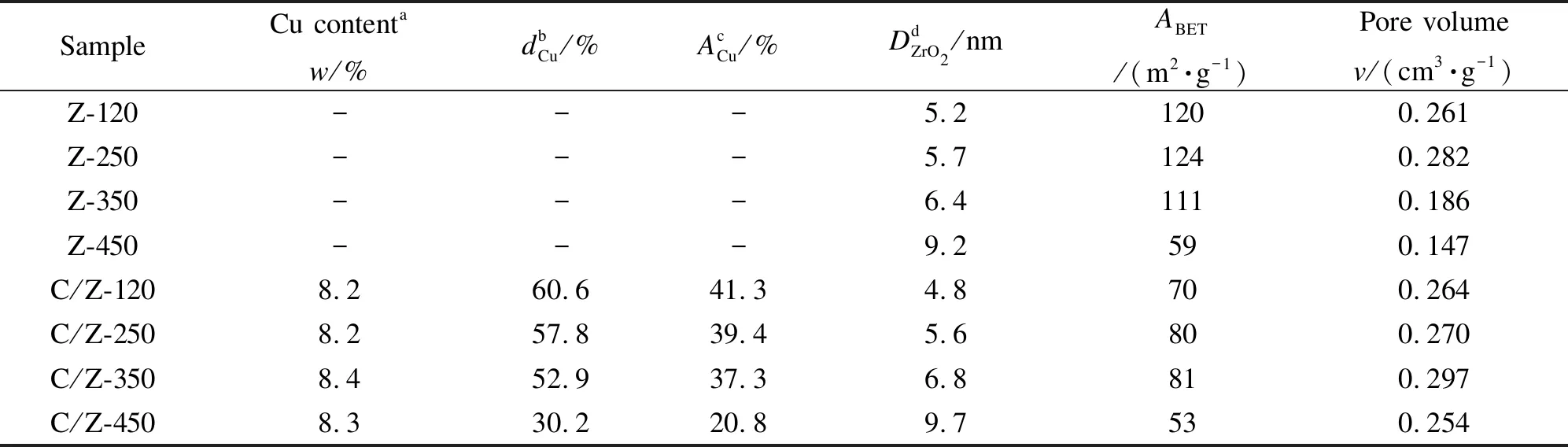
表 1 经不同温度焙烧的ZrO2载体及相应CuO/ZrO2催化剂的结构性质
a: measured by ICP-OES;b: Cu dispersion calculated from N2O titration results;c: Cu metal area calculated from N2O titration results;d: crystallite size ofm-ZrO2calculated by the Scherrer equation using the diffraction at 2θ=32.8°
为进一步研究样品的CuO分散状态,采用N2O滴定法测得了系列CuO/ZrO2催化剂的Cu分散度和Cu金属比表面积,结果见表1。由表1可知,样品的Cu分散度和Cu金属比表面积均随着ZrO2焙烧温度的升高而降低,这与各样品中高分散型态CuO的含量变化规律相一致。值得注意的是,样品C/Z-450的Cu分散度和Cu金属比表面积相较于样品C/Z-120降低幅度最为明显,均仅为后者的50%,这可能与载体Z-450的晶粒粒径明显较大有关。图2显示了ZrO2焙烧温度、ZrO2载体晶粒粒径以及CuO/ZrO2催化剂的Cu分散度三者之间的关系。由图2可知,随着ZrO2焙烧温度的升高,Cu分散度与ZrO2载体的晶粒粒径呈现完全相反的变化趋势,这一结果直接表明,载体ZrO2的晶粒粒径制约着CuO的分散状态,即ZrO2载体晶粒粒径越小,相应催化剂上的Cu分散度就越高。类似的现象也出现在Au/ZrO2[28]、CuO/Ce1-xTixO2[30]等负载型金属催化剂体系中。原因是小粒径氧化物晶粒表面会具有较多的配位不饱和金属离子(与氧空位成对共存)等缺陷位点[31],这些位点通常是负载金属的沉积或成核中心[32]。
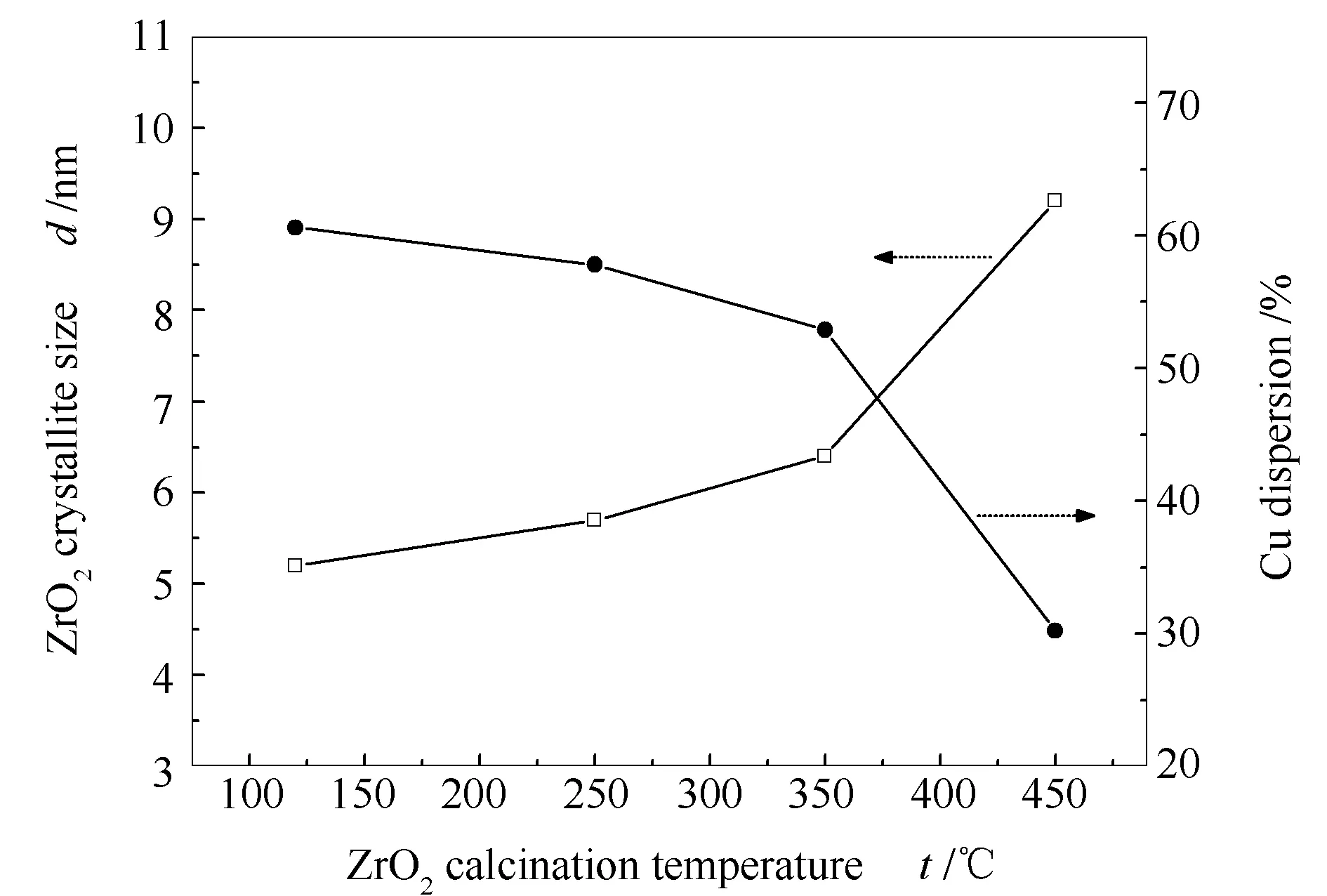
图 2 ZrO2焙烧温度与ZrO2载体晶粒粒径及催化剂Cu分散度间的关系
鉴于载体的织构性能同样会影响到CuO的负载和分散,作者对经不同温度焙烧的ZrO2载体进行了N2物理吸附-脱附实验,结果见图3(a)、图3(b)。由图3 (a)可知,各ZrO2载体的吸附-脱附等温线均为Ⅳ型曲线(BDDT分类),在相对压力p/p0> 0.7的滞后环为H1型,反映的是规则有序的颗粒堆积孔[33]。由图3(b)可知,各ZrO2载体均呈现双孔分布的孔结构:孔径小于5 nm的孔为ZrO2颗粒内部由纳米晶粒堆积形成的一次孔[26];孔径在5-100 nm的孔为ZrO2颗粒相互堆积形成二次孔,对应图3(a)中的H1型滞后环。焙烧温度≤ 350 ℃时,随着焙烧温度的升高,ZrO2颗粒内的一次孔逐渐增多,说明在适当温度下焙烧有助于ZrO2颗粒内部孔道的形成。焙烧温度为450 ℃时,ZrO2一次孔的最可几孔径明显增大,孔分布更为集中,表明ZrO2颗粒内部孔道发生明显重构,这应该与其晶粒粒径显著长大相关。此外,由图3(b)还可知,随着焙烧温度的升高,ZrO2颗粒间的二次孔逐渐减少,最可几孔径也逐渐渐小,计算得到的BET比表面积和总孔容积(见表1)也总体呈现减小趋势,说明升高焙烧温度会导致ZrO2颗粒团聚或烧结。ZrO2载体的这一结构变化必然不利于CuO在其表面的分散,这与XRD和N2O滴定实验结果相吻合。
图3(c)、图3(d)为系列CuO/ZrO2催化剂的N2物理吸附-脱附实验结果。由图3(c)可知,相比于ZrO2载体,各催化剂样品的H1型滞后环均明显变窄并向相对压力较高的区域发生偏移,说明各样品的二次孔尺寸有所增大。观察图3(d)并对比图3(b)可以看出,各催化剂样品的的二次孔最可几孔径均约为50 nm,并大于相应的ZrO2载体。而且各样品的二次孔数目也基本相当,且均多于相应的ZrO2载体。上述负载CuO后产生的扩孔和增孔现象可能源于晶相CuO对ZrO2颗粒的阻隔作用,该作用抑制了ZrO2颗粒间的团聚。样品C/Z-450中晶相CuO含量最高,上述抑制效果也最为明显。这种阻隔作用通过对比ZrO2及相应CuO/ZrO2催化剂的总孔容积(见表1)也可以看出。此外,对比图3(d)和图3(b)还可发现,各CuO/ZrO2样品的一次孔数目相比于ZrO2载体均有所减少,这可能是由于负载的CuO堵塞了ZrO2颗粒表面的部分一次孔。这也使得各CuO/ZrO2样品的BET比表面积相较于ZrO2载体均明显减小。
2.2 CuO/ZrO2催化剂的还原性能
图4为系列CuO/ZrO2催化剂的H2-TPR谱图。
图中各样品均出现α、β和γ三个还原峰。前期研究[26,29]对三个还原峰的归属如下:α峰归属为表面高分散CuO簇的还原;β峰归属为高分散Cu-[O]-Zr物种的还原;γ峰归属为晶相CuO的还原。为了更准确研究催化剂中各CuO物种的相对含量及还原能力,表2为各样品的H2-TPR谱图拟合结果(采用Gauss-Lorentz法),包括各拟合峰的峰温和其所对应的耗氢量。

表 2 不同温度焙烧的ZrO2为载体的CuO/ZrO2催化剂的还原性能
a: values in parentheses are the H2consumption calculated by fitting results;b: calculated from the H2signal peak (m/z= 2) area in CO-TPR-MS profiles by assuming the relative concentration of active OH in C/Z-250 is 1.00
由图4及表2可知,ZrO2焙烧温度对CuO/ZrO2催化剂中三种CuO物种的含量及其还原能力有重要影响。各催化剂中晶相CuO(γ物种)的含量随ZrO2载体焙烧温度的升高而升高,这与上文的XRD表征结果相一致。相反,高分散CuO物种(α物种和β物种)的含量总和则随ZrO2载体焙烧温度的升高而降低,这与N2O滴定实验测定的Cu分散度和Cu金属比表面积变化规律相一致,也暗示Cu分散度和Cu金属比表面积主要由高分散CuO物种贡献。对于高分散CuO簇(α物种),其含量随ZrO2焙烧温度的升高先略微升高而后降低,这与ZrO2载体的BET比表面积(见表1)变化趋势一致,说明CuO簇主要分布在ZrO2的表面。而Cu-[O]-Zr物种(β物种)的含量则按如下顺序依次降低:C/Z-120 > C/Z-250 > C/Z-350 > C/Z-450,这与ZrO2载体的晶粒粒径随焙烧温度的变化规律恰好相反,说明ZrO2载体的晶粒粒径可能是影响Cu-[O]-Zr物种含量的主要因素。而由ZrO2晶粒粒径所决定的表面缺陷位浓度(如表面氧空位浓度)可能是制约Cu-[O]-Zr物种含量的内在因素。
结合图4及表2中各还原峰峰温数据,可以发现α峰及β峰峰温随ZrO2焙烧温度的升高逐渐降低,说明系列样品中高分散CuO簇及Cu-[O]-Zr物种的还原能力按如下顺序依次减弱:C/Z-450 > C/Z-350 > C/Z-250 > C/Z-120。对于载体表面的高分散CuO物种,其还原能力与其和载体间的结合力(作用力)有关,通常结合力越强,这类CuO就越难还原[34]。而表面羟基作为金属氧化物的重要表面特征,对负载金属与金属氧化物之间的结合力有重要影响[35]。为了分析ZrO2载体表面羟基与高分散CuO物种的还原能力是否相关,作者通过H2O程序升温脱附-质谱跟踪实验(H2O-TPD-MS)对各ZrO2载体表面羟基浓度进行了研究,结果见图5。由图5可知,30-230 ℃的H2O(气体)脱附峰(标记为A峰)归属为ZrO2表面吸附水的受热脱附[36],脱附温度高于230 ℃的H2O(气体)脱附峰(标记为B峰)归属为ZrO2表面羟基的受热脱附[36]。由B峰面积可知,各ZrO2载体的表面羟基浓度顺序如下:Z-450 < Z-350 < Z-250 < Z-120,这与催化剂中高分散CuO物种(α及β物种)的还原温度高低顺序相同,说明ZrO2载体的表面羟基可能有助于增强催化剂上高分散CuO物种与ZrO2之间的结合力,从而降低高分散CuO物种的还原能力。载体Z-120的表面羟基浓度最高,相应的,样品C/Z-120上的α及βCuO物种的还原温度也最高,即还原能力最弱。上述结果与XRD结果所表明的ZrO2载体焙烧温度较低时CuO-ZrO2间相互作用会增强的现象相吻合。
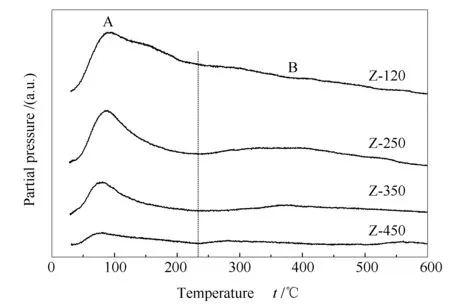
图 5 不同温度焙烧的ZrO2的H2O-TPD质谱谱图
对于晶相CuO,其还原温度并未随ZrO2载体表面羟基浓度的降低(即焙烧温度的升高)而降低,相反却有所升高。这说明晶相CuO与ZrO2之间的相互作用受ZrO2载体表面羟基的影响较小。晶粒粒径存在差异可能是导致上述晶相CuO还原能力不同的主要原因。
为进一步研究CuO/ZrO2催化剂中各种含氧物种的还原性能,对各样品进行了CO-TPR-MS表征,结果见图6。
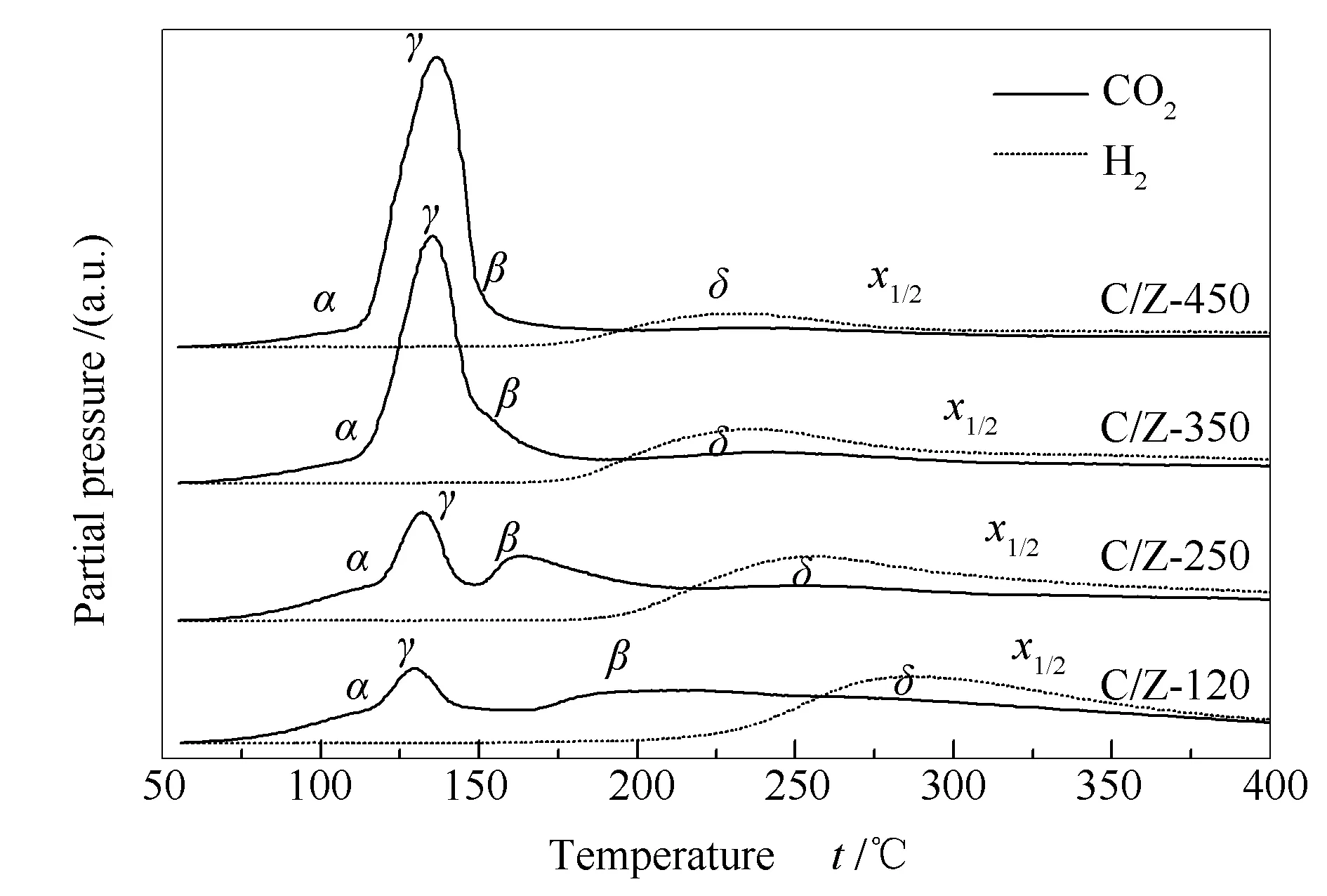
图 6 不同温度焙烧的ZrO2为载体的 CuO/ZrO2催化剂的CO-TPR-MS谱图Figure 6 CO-TPR-MS profiles of the CuO/ZrO2catalysts prepared with the ZrO2 supports calcined at different temperatures(CO2, m/z = 44; H2, m/z = 2)
使用CO-TPR-MS技术不仅可以根据式(6)测定催化剂中各CuO物种的还原能力和含量,还可根据式(7)测定催化剂活性表面羟基的还原能力和含量[37,38]。与H2-TPR还原峰归属一致,CO-TPR-MS图中α、β和γCO2(m/z= 44)信号峰也分别归属为ZrO2表面高分散CuO簇的还原、高分散Cu-[O]-Zr物种的还原和晶相CuO的还原[26,29]。根据式(7),δCO2信号峰伴随着H2的生成,归属为活性表面羟基的还原,即CuO/ZrO2催化剂活性表面羟基所参与的WGS反应(简称表面WGS反应)。
CuO + CO → Cu + CO2
(6)
OH(ZrO2)+ CO → CO2+ 1/2H2
(7)
由图6可知,随着ZrO2载体焙烧温度的升高,γCO2信号峰的峰面积逐渐增大,而α峰及β峰的峰面积则逐渐减小,表明晶相CuO的含量逐渐升高,而高分散CuO簇和Cu-[O]-Zr物种的含量则逐渐减少。此外,β峰的峰温随ZrO2载体焙烧温度的升高逐渐降低,表明Cu-[O]-Zr物种的还原能力逐渐增强。上述结果均与H2-TPR表征结果一致。如本文引言部分所述,前期研究已证实[29],上述三种CuO物种中,仅有Cu-[O]-Zr物种是CuO/ZrO2催化剂上催化WGS反应的活性CuO物种。因此,通过改变载体的焙烧温度,诱导Cu-[O]-Zr物种数量和还原能力发生改变,必然会对CuO/ZrO2催化剂的WGS反应催化活性产生影响。
从图6和表2还可知,ZrO2载体的焙烧温度越高,δ峰的峰温就越低,说明催化剂活性表面羟基的还原能力越强,即催化剂上越容易发生如式(7)所示的表面WGS反应。其中,样品C/Z-450的δ峰峰温较样品C/Z-120低约50 ℃,说明该催化剂上WGS反应发生的起活温度较后者低约50 ℃。值得注意的是,改变载体焙烧温度,β峰与δ峰具有相同的偏移方向和相近的偏移程度,这说明Cu-[O]-Zr物种的还原能力直接制约着催化剂活性表面羟基的还原能力。仔细观察图6中各样品的质谱谱线均可以发现,仅当Cu-[O]-Zr物种被还原后,活性表面羟基才开始被还原,即CO才能与活性羟基发生表面WGS反应,这也再次表明Cu-[O]-Zr物种(而不是α或γ物种)才是对WGS反应具有催化作用的CuO物种。此外,通过计算H2(m/z= 2)谱线下的峰面积(反映H2的生成量),根据式(7)可以推算出各催化剂中活性表面羟基的相对含量,结果见表2。由表2可知,样品C/Z-250中活性表面羟基数量最多,其次是样品C/Z-350和C/Z-120,样品C/Z-450上的活性羟基数量最少。活性表面羟基的多少,同样会影响CuO/ZrO2催化剂的WGS反应催化活性。
2.3 催化剂的WGS活性
图7为系列CuO/ZrO2催化剂的WGS催化活性。
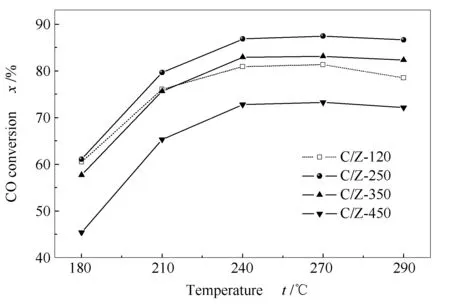
图 7 不同温度焙烧的ZrO2为载体的CuO/ZrO2催化剂的WGS活性
由图7可知,在整个测试温区(180-300 ℃) 内,随ZrO2载体焙烧温度的升高,CuO/ZrO2催化剂的催化活性均呈现先升高后降低的“火山型”变化趋势。其中,样品C/Z-250的催化活性最高,而C/Z-450的催化活性最低。样品C/Z-250在反应温度为270 ℃时的CO转化率达到最高值87.5%,较C/Z-450提高了20%。此外,各样品的催化活性还显著高于相同测试条件下CuO/ZnO和CuO/CeO2的催化活性[26]。
联系前面的表征结果,改变ZrO2载体的焙烧温度,CuO/ZrO2催化剂的WGS活性产生差异的原因可能如下:
对于样品C/Z-250、C/Z-350及C/Z-450,其WGS催化活性可以与表面Cu-[O]-Zr物种的数量很好的关联:即Cu-[O]-Zr物种数量越多,相应样品的WGS催化活性就越高。根据CO-TPR-MS表征结果,Cu-[O]-Zr物种数量的增多,意味着催化剂表面活性位点的增多,相应地,样品的催化活性就越高。需要强调的是,CuO/ZrO2催化剂表面Cu-[O]-Zr物种的数量与ZrO2载体的晶粒粒径直接相关。ZrO2载体的晶粒粒径越小,表面Cu-[O]-Zr物种数量就越多。这是改变ZrO2载体焙烧温度会影响CuO/ZrO2催化剂WGS催化活性的第一种原因。
对于样品C/Z-120,尽管其Cu分散度最高,Cu金属比表面积最大,并具有最多的表面Cu-[O]-Zr物种,其WGS催化活性却不是最高的。这应该与其表面Cu-[O]-Zr物种的还原能力较弱有关。CO-TPR-MS结果表明,催化剂活性表面羟基的还原能力(即活性表面羟基与CO发生表面WGS反应的能力)与Cu-[O]-Zr物种的还原能力密切相关。Cu-[O]-Zr物种越容易被还原,表面WGS反应就越容易被触发。由H2-TPR和H2O-TPD实验结果可知,焙烧温度决定了ZrO2载体表面羟基(注意与CuO/ZrO2催化剂活性表面羟基加以区分)的含量,从而影响到Cu-[O]-Zr物种的还原能力,并最终对CuO/ZrO2催化剂的WGS催化性能产生影响。这是改变ZrO2载体焙烧温度会影响CuO/ZrO2催化剂WGS催化活性的第二种原因。
此外,CuO/ZrO2催化剂的WGS活性还可以与其活性表面羟基的数量(见表2)较好的关联,这显示了活性表面羟基对WGS反应的重要性。暗示CuO/ZrO2催化剂上的WGS反应机理可能遵循了有活性表面羟基参与的表面甲酸盐机理[17,25]。
3 结 论
本研究采用沉积-沉淀法制备了系列高活性CuO/ZrO2WGS催化剂。通过改变ZrO2载体的焙烧温度实现了对CuO/ZrO2催化剂上活性Cu-[O]-Zr物种的含量及还原性能的调变。一方面,提高ZrO2的焙烧温度会使其晶粒粒径增大,这会导致CuO/ZrO2催化剂Cu分散度降低,Cu金属比表面积减小,特别是活性Cu-[O]-Zr物种含量降低;另一方面,高温焙烧的ZrO2载体其表面羟基浓度显著减小,这使得CuO-ZrO2之间的结合力变弱,Cu-[O]-Zr物种较易被还原。相应地,CuO/ZrO2催化剂上活性表面羟基的还原能力也明显增强,即活性表面羟基与CO发生WGS反应的起始温度显著降低。改变ZrO2焙烧温度所产生的上述两方面的影响使得样品C/Z-250(中等温度焙烧)表现出最佳的WGS催化活性。
