在地钱中构建简便、高效的基因表达检测体系
2019-04-13苗志奇徐美慧
苗志奇,周 璐,徐美慧
在地钱中构建简便、高效的基因表达检测体系
苗志奇,周 璐,徐美慧
(上海交通大学农业与生物学院,上海 200240)
实时荧光定量聚合酶链式反应(qRT-PCR)广泛应用于基因转录水平的差异分析.选择稳定表达的参比基因,设计理想的参比引物扩增参比基因进而对样本进行标准化处理是得到可靠的qRT-PCR分析结果的前提.地钱具有基因组小、生长周期短、易于培养繁殖、转基因效率高等优势,已成为一种新兴的合成生物学模式植物.植物合成生物学的研究产生了大量的转基因和突变系等地钱材料,同时也提出了在地钱底盘中提升qRT-PCR的效率和可靠性,从而快速准确分析外源以及内源基因表达水平的现实需求.对地钱基因组进行生物信息学分析发现,地钱中参比基因ACT7存在两个高度同源的基因序列,其中任一基因的表达均不太稳定,但两个基因的表达存在一定的互补性.为了在地钱中构建简易、高效的qRT-PCR分析体系,利用数学模型,分析qRT-PCR误差传导机制,建立参比引物的综合评价指标体系.研究发现,能够同时扩增两个ACT7高度同源基因的共退火引物对应的表达水平更为稳定,其中编号为3~6的引物在扩增效率、表达水平及稳定性等多个指标上表现均衡优越,从而开发了一种在地钱中快速准确检测基因表达水平的qRT-PCR平台技术,同时也为在其他物种中如何优化qRT-PCR检测体系指明方向.
实时荧光定量PCR;地钱;ACTIN参比基因;共引物设计
实时荧光定量PCR(real time fluorescence quantitative PCR,qRT-PCR)是在转录水平上分析基因表达水平的主流检测技术,具有灵敏度高、特异性强、重复性好的优点[1].由于检测样本在RNA浓度、质量和反转录效率上可能存在差异,选择合适的参比基因,进而标准化处理,消除样品间的差异是保证qRT-PCR结果准确可靠的关键.理想的参比基因要求在各类组织以及不同生长发育阶段均稳定表达,表达水平不受外源胁迫因素的影响,同时其表达水平与目标基因表达水平相近[2].常用的植物参比基因有甘油醛-3-磷酸-脱氢酶基因(glyceraldehyde-3-phosphatede-hydrogenase,GAPDH)、肌动蛋白基因(actin)、微管蛋白基因(tubulin)、18SrRNA和28SrRNA等[3].研究发现上述参比基因在不同物种的表达水平及其稳定性存在差异,针对不同物种进行综合评价选择合适的参比基因就显得尤为重要[4].
地钱()是苔类植物,具有基因组小、生长周期短、易于培养繁殖、转基因效率高等优势[5].和大多数高等植物不同,地钱的孢子体世代为单倍体植株,不存在多倍体植物共有的基因杂合问题,可为基因功能研究提供更为简便高效的研究平台.除常规的有性繁殖外,地钱具有和微生物类似的胚芽无性生殖方式,每个胚芽都起源于单细胞,可以发育成完整的地钱个体.采用这种独一无二的胚芽繁殖方式可以快速获得大量和亲本遗传背景完全一致的地钱植株,因此利用地钱开展研究工作可以有效减少背景噪音对研究目标的干扰,提升研究结果的可靠性.越来越多的研究者在地钱平台上开展研究工作,包括正己醛和己烯醛的生物合成[6]、羟化类胡萝卜素(叶黄素)的生物合成[7]、丝氨酸生物合成酶和3-磷酸甘油酸脱氢酶的生化特征研究等[8].近年来,根癌农杆菌介导的高效地钱遗传转化体系逐渐建立,利用胚芽繁殖方式的单克隆特性,成功解决了植物转基因过程中常见的嵌合体问题.使得地钱逐渐发展成植物合成生物学的经典底盘[9].
随着植物合成生物学研究的开展,在地钱底盘上构建全新的生物通路,赋予地钱全新的功能成为研究多细胞生物发育以及生物制造的热点之一.植物合成生物学研究产生了大量的转基因和突变系等地钱材料[10],同时也提出了在地钱底盘中提升qRT-PCR的效率和可靠性,从而快速准确分析外源以及内源基因表达水平的现实需求.目前地钱底盘中广泛应用的qRT-PCR参比基因有MpACT、MpAPT、MpGAPC1、MpPEX和MpUBQ10等.研究发现MpACT基因在不同生长发育阶段以及激素处理下均具有良好的稳定性,但在非生物胁迫处理下稳定性差,因此建议使用MpACT和MpAPT作为双参比基因对检测数据进行标准化校正[11],提高基因表达水平分析的准确性,但双参比体系需要设计两对参比引物分别进行扩增,增加了实验设计的复杂性,同时也降低了分析效率.
本文首先建立了qRT-PCR的数学模型,分析基因表达水平检测过程中的误差传导规律,从而指导基因表达水平的精确测量与体系的综合评价.而后对地钱进行基因组数据进行分析,发现常用参比基因ACT7在地钱中存在两个高度同源的基因序列,且其表达存在一定的互补性,推测参比基因ACT7的多高度同源的基因序列是导致其在地钱底盘中稳定性差的重要因素之一.进而设计系列参比引物构建qRT-PCR体系,采用一对参比引物,分别或同时扩增参比基因ACT7的不同高度同源的基因序列,通过综合指标分析确定最佳参比引物,进而建立一种简便、稳定的qRT-PCR检测体系,满足在地钱底盘上快速、高效检测基因表达水平的需求,同时也为其他物种中如何优化qRT-PCR检测体系指明方向.
1 材料与方法
1.1 植物材料与培养条件
本研究所用野生型地钱(M-w)为雌性粗裂地钱FSN-1,保存于上海交通大学植物生物技术中心.
研究所用转基因地钱材料为两组转基因株系(M-tp+;M-tp-).其共同特点是强表达法尼基焦磷酸合成酶(farnesyl diphosphate synthase,FPS)与广藿香合成酶(patchoulol synthase,PTS),差别是通过信号肽将外源蛋白分别定位于细胞质和质体中,实现广藿香的异源合成(详细的载体构建、转基因以及含量检测将单独成文发表).上述外源基因的转入理论上对ACT7基因的表达不产生影响.
选取地钱胚芽接种在含1%琼脂的1/2B5培养基上,培养箱温度为25℃,光周期为16h光照(白光,10000lx)/8h黑暗[9].培养周期为30d,即可获得成熟的地钱组织材料.本研究材料是在培养15d后,采集地钱叶状体组织后液氮速冻,-80℃贮藏备用.
1.2 总RNA的提取
按照TIANGEN RNAprep Pure Plant Kit试剂盒操作方法,提取各样品的总RNA.获得的RNA产物检验其完整度(见图1),并测量浓度后于-80℃贮藏 备用.
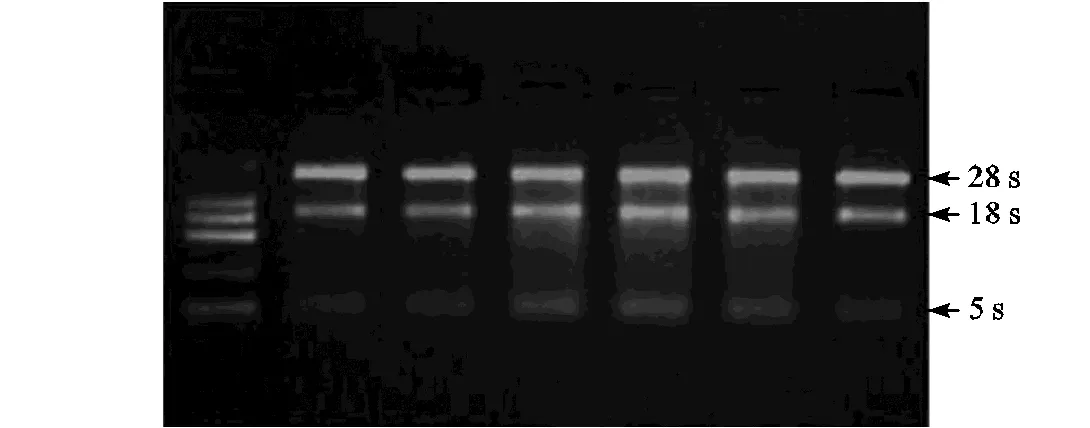
图1 总RNA质量电泳图
1.3 反转录合成cDNA
按照PrimeScriptTM RT Reagent Kit(Perfect Real Time)试剂盒(TaKaRa公司)操作方法,将各样品总RNA反转录合成cDNA第一链.获得的cDNA产物直接用于PCR或-20℃贮藏备用.
1.4 PCR引物设计
用ACT7基因序列(U27811.1)对地钱基因组(GCA_001641455.1)进行BLAST分析,发现有两个高度同源的基因序列,分别命名为ACT7-1与ACT7-2.采用CLUSTW对两个同源ACT基因进行序列比对,分析发现序列一致区域和差异区域.采用Oligo 7.0软件,针对差异区域设计分别与ACT7-1、ACT-2特异性退火的特异引物;针对序列一致区域,设计能同时和ACT7-1、ACT-2结合的共退火引物.引物序列见表1.引物合成由生工生物工程股份有限公司 完成.
1.5 参比基因的荧光定量PCR分析
定量PCR体系总反应体积为20μL,包括10μmol/L 正向和反向引物各0.6μL、RNase-Free ddH2O 5.8μL和TIANGEN SuperReal PreMix Plus(SYBR Green)10μL,以及25倍稀释的待测样品cDNA 3μL.
表1 地钱中参比基因qRT-PCR的引物序列

Tab.1 qRT-PCR candidate reference genes’ primers in Marchantia polymorpha
荧光定量专用反应管为Axygen公司的PCR-0108-LP-RT-W,采用Roche公司lightcycle®96荧光定量PCR以进行.PCR反应条件为:95℃预变性900s,然后进行40个循环的PCR扩增,每个循环包括95℃变性20s,55℃退火20s,72℃延伸20s 3步.每个样品设置3个重复组进行测量.
1.6 数据处理与分析
1.6.1 扩增效率与扩增稳定性
理想PCR过程的扩增效率为100%,即每经过一轮PCR扩增,目标序列的数量实现翻倍增长.在实际实验中,选择不同引物导致的退火效率和二级结构的差异会使扩增效率偏离理想值.

(1)
扩增稳定性定义如下:

(2)
1.6.2 表达水平与表达稳定性
基因的表达水平可以用等量cDNA样本中目标转录本的拷贝数0来表示:

(3)
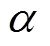
表达稳定性定义如下:

(4)
可见表达水平离散系数越小,说明表达水平的稳定性越好.
1.6.3 表达水平的最佳估计值与可信度
针对同一参比基因设计多对引物,分别进行qRT-PCR,可以获得表征该参比基因表达水平多个测量值0i(=1,2,3,…).表达水平的最佳估计值定义为各测量值按照其准确度的加权平均.

(5)
式中准确度E为第个表达水平测量值误差的倒数,表达式为
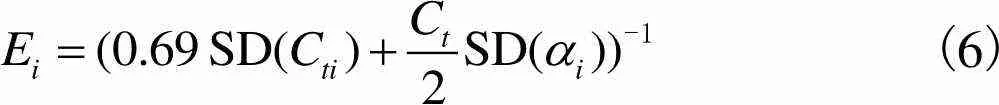
(6)
式中SD为标准差,误差的详细推导过程见第2.1节.
引物的可信度定义如下:

(7)
按照第对引物测量获得的参比基因表达水平0i和参比基因的最佳估计值越接近,则说明第对引物的可信度指标越高.
1.6.4 DNA污染检测能力分析
作为qRT-PCR模板的cDNA可能含有基因组DNA污染,理想的引物设计能够直接检测是否存在基因组DNA污染.因此定义引物的DNA污染检测能力T如下:
若引物可区分cDNA样品中是否有DNA污染,则T=1;否则T=0.
引物的DNA污染检测能力可以用参比基因DNA中引物扩增区域是否存在内含子来表征.若在基因组DNA序列上参比引物退火位点之间含有内含子序列,则以cDNA和可能污染的基因组DNA作为模板进行PCR会扩增产生不同长度的片段,使溶解度曲线从标准的单峰曲线变成明显的双峰曲线,从而判断基因组DNA污染的存在.
1.6.5 数据标准化处理与综合分析
在采用多指标综合分析引物优劣时,为避免各指标变化幅度的巨大差异对分析结果产生误导,需要对各指标数据进行标准化处理,使之处于同一数量级,提高其可比性.
本文采用的归一化方法是min-max标准化(min-max normalization),转换函数如下:

(8)
式中:下标max为样本数据的最大值;下标min为样本数据的最小值.标准化结果将所有指标数据映射到0~1之间[12]. 采用R语言绘制星图,对各引物进行综合分析.
2 结果与讨论
2.1 qRT-PCR数学模型与误差传递分析
在qRT-PCR扩增前期,引物、核苷酸底物充足,该过程中荧光信号强度随扩增循环数的增加呈现指数增长,如下式所示:
因此,样本中目标转录本的拷贝数0为
取对数,得
根据全微分公式:
2.2 参比基因的选择与引物的设计
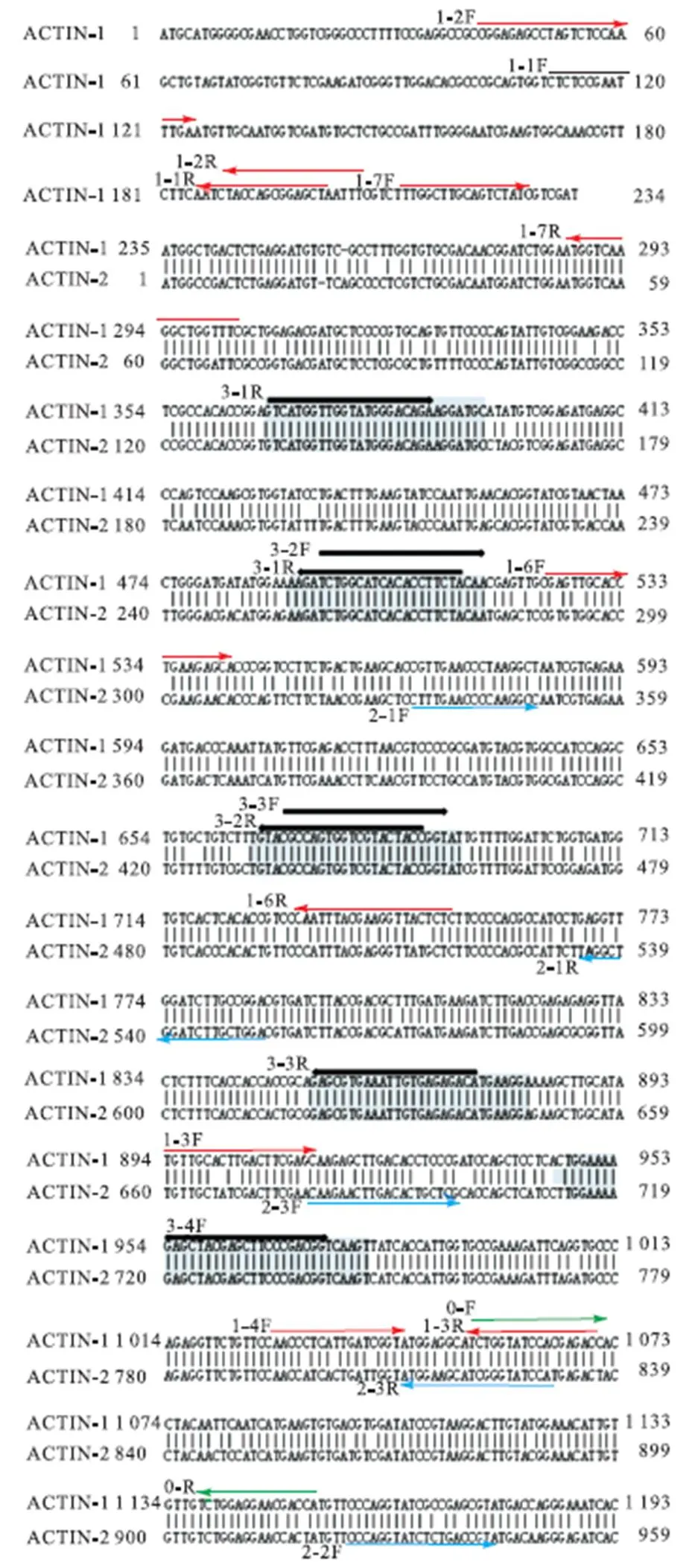
对比地钱基因组数据,发现地钱中存在两个ACT7同源基因:ACT7-1与ACT7-2.ACT7[11](1134bp)与ACT7-1(1368bp)的235~1368bp完全相同,与ACT7-2(1134bp)的相似性高达89%. 如图2所示:红色箭头表示扩增ACTIN-1的上下游引物;蓝色箭头表示扩增ACTIN-2的上下游引物;黑色箭头表示同时扩增ACTIN-1和ACTIN-2的上下游引物;绿色箭头为文献中已发表的常用引物-0的上下游引物;所有引物的编号均位于箭头左端;灰色表示ACTIN-1和ACTIN-2完全相同的部分.
NCBI Primer-BLAST分析发现文献[11]中扩增ACT7基因的引物(编号为0,正向引物为:5’-AGGCATCTGGTATCCACGAG-3’;反向引物为:5’-ACATGGTCGTTCCTCCAGAC-3’)与ACT7-1能实现精准退火,从而在PCR过程中进行高效扩增;但该对引物与ACT7-2存在多于4个碱基的差异,难以退火,不能在PCR过程中实现有效扩增.推测地钱中ACT7的高度同源的基因序列以及引物退火的特异性是造成以ACTIN为参比基因的地钱qRT-PCR分析不稳定的重要原因.
在序列比对的基础上,分别针对两个ACT7同源基因的序列一致区域设计共退火引物,针对序列差异区域设计基因特异引物(如表1所示),分别考察这两个ACT7同源基因单独或共同作为参比基因的可行性.
首先选择0号引物扩增ACT7-1,2-1引物扩增ACT7-2,检测野生型地钱以及两种不同广藿香转基因地钱中两个ACT7同源基因的表达.ACT7-1在各株系表达水平的离散系数为0.24,ACT7-2在各株系表达水平的离散系数为0.23,两个ACT7同源基因的表达并不稳定,和Saintmarcoux等[11]的报道相一致.但两个同源ACT7基因的表达存在明显的互补关系,二者表达量之和的离散系数为0.05,远比其中任何一个都更为稳定,如图3所示.以上现象表明,用两个ACT7同源基因共同作为参比基因,可望解决ACTIN参比基因在地钱底盘中表达不稳定问题.
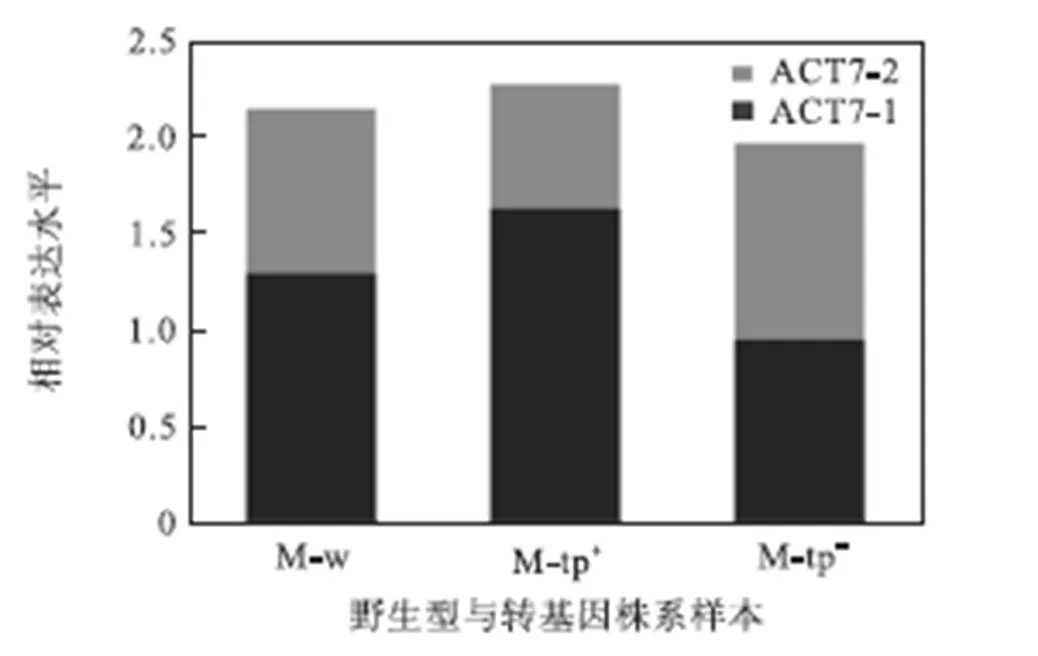
图3 ACT7-1与ACT7-2相对表达水平关系
2.3 基因共退火引物与特异引物对qRT-PCR稳定性的影响分析
针对表1中ACT7-1基因特异引物、ACT7-2基因特异引物以及共退火引物3组不同ACTIN参比引物,分别构建qRT-PCR体系,检测野生型地钱以及两种转基因地钱中ACTIN基因的表达,从扩增效率、扩增稳定性、表达水平以及表达稳定性这4个指标来考察不同类型引物对qRT-PCR体系稳定性的影响.

从表达水平(见图6)分析发现,不同地钱组别引物对应ACTIN基因表达水平存在明显差异.方差分析对应<0.001,也表明3组数据存在极显著差异.共退火引物组的ACTIN基因表达水平的平均值最高,且相较于其他两组,偏离数据少,较为稳定. ACT7-1的表达水平高于ACT7-2,且分布更为集中.

图4 3组不同退火特异性引物对应的PCR扩增效率 分析

图5 3组不同退火特异性引物对应的PCR扩增稳定性分析

图6 3组不同退火特异性引物对应的参比基因表达水平分析
根据qRT-PCR误差传导规律,扩增效率的误差水平是基因表达稳定性的决定因素,与图7中表达稳定性分析结果完全吻合.共退火引物组对应的表达稳定性最好,y=0.962,远高于两个基因特异引物组的0.40和0.58,说明利用共退火引物扩增参比基因获得的表达水平更为稳定.基因表达的稳定性数值只有扩增稳定性的1/10左右,说明扩增误差被放大了10倍左右形成最终的基因表达误差.而临界循环误差在总误差中所占比例几乎可以忽略.
综上所述,共退火引物组的扩增效率和其他两组基因特异引物无明显区别,但扩增稳定性、参比基因表达水平及稳定性明显优于另外两组基因特异引物.后续将针对共退火引物,采用多种评价指标进行系统分析,以比较确认最佳的共退火引物,提升qRT-PCR的可靠性和准确度.

图7 3组不同退火特异性引物对应的参比基因表达稳定性分析
2.4 共退火ACT7引物的综合性能分析
以文献[11]中设计的0号引物为对照,针对表1中6对ACT7共退火引物,对10株野生型地钱以及20株广藿香转基因地钱进行qRT-PCR分析,从扩增效率、扩增稳定性、表达水平、表达稳定性、可信度与能否检验DNA污染这6个方面进行综合考察.在min-max标准化处理后,绘制星图(见图8)进行综合分析.
从单指标分析看,星图中某个指标对应轴越长,说明该指标性能越好.图8显示从单个指标分析看,引物3-4扩增效率指标最佳,接近于理想值1;从扩增稳定性指标看,最优的引物为3-5,对应扩增效率最稳定;在所有引物中,引物3-6对应的表达水平最高,以其为基础构建qRT-PCR体系可获得最高灵敏度和更低的检测下限;单从表达稳定性分析,最优的引物应为3-5;可信度分析的最优引物为3-6;引物3-3和3-6以基因组DNA为模板进行PCR扩增时,由于扩增区产物包含内含子,扩增出的条带长度大于以cDNA为模板的条带(见图9),也会明显改变产物的溶解度曲线,能够区分cDNA样品中是否有DNA 污染.

A—扩增效率理想度;B—扩增稳定性;C—表达水平;D—表达稳定性;E—可信度;F—检验DNA污染
从多指标综合分析看,星图对应面积越大,畸形度越小,对称度越高,说明对应引物综合性能越好,从图8可以看出引物3-6在6个指标中表现最为均衡,在数据标准化后扩增效率为0.45,扩增稳定性为0.73,表达水平为1.00,表达稳定性为0.49,可信度为1.00,检验DNA污染为1.00,故而引物3-6是地钱最佳的ACTIN共退火参比引物.

图9 ACTIN共退火引物的退火位置示意(单位:bp)
3 结 语
地钱中常用的qRT-PCR参比基因ACT7有两个高度同源的基因序列,且表达水平呈现明显的互补特征,以其中任何一个为参比基因都存在参比基因的不稳定问题,影响qRT-PCR结构的准确性.通过设计共退火引物,将两个ACT7同源基因一起作为参比基因使用,在不增加系统复杂性的同时,极大地提高了ACT7参比基因的稳定性,提升了基因表达分析的可靠性.在数学建模的基础上,建立了引物多指标综合分析体系,找到了最佳共退火引物3-6,建立了适合在地钱底盘中基因表达分析的高效、可靠的qRT-PCR技术,为其他物种中如何优化qRT-PCR体系提高基因表达数据的可靠性提供了有益借鉴.
[1] 董恩妮,梁 青,李 利,等. 实时荧光定量PCR内参基因的选择[J]. 中国畜牧杂志,2013,49(11):92-96.
Dong Enni,Liang Qing,Li Li,et al. Selection of real-time quantitative PCR internal reference genes [J]. Chinese Journal of Animal Husbandry,2013,49(11):92-96(in Chinese).
[2] 胡瑞波,范成明,傅永福. 植物实时荧光定量PCR内参基因的选择[J]. 中国农业科技导报,2009,11(6):30-36.
Hu Ruibo,Fan Chengming,Fu Yongfu. Selection of internal reference genes in real-time fluorescent quantitative PCR for plants[J]. China Agricultural Science and Technology Guide,2009,11(6):30-36(in Chinese).
[3] 孙美莲,王云生,杨冬青,等. 茶树实时荧光定量PCR分析中内参基因的选择[J]. 植物学报,2010,45(5):579-587.
Sun Meilian,Wang Yunsheng,Yang Dongqing,et al. Selection of internal reference genes in real-time PCR analysis of tea plants[J]. Plant Journal,2010,45(5):579-587(in Chinese).
[4] 袁 伟,万红建,杨悦俭. 植物实时荧光定量PCR内参基因的特点及选择[J]. 植物学报,2012,47(4):427-436.
Yuan Wei,Wan Hongjian,Yang Yuejian. Characteristics and selection of the internal reference gene of real-time fluorescent quantitative PCR for plants[J]. Journal of Plant Science,2012,47(4):427-436(in Chinese).
[5] Tsuboyama S,Kodama Y. AgarTrap:A simplified Agrobacterium-mediated transformation method for sporelings of the liverwortL. [J]. Plant & Cell Physiology,2014,55(1):229-236.
[6] Tawfik M M,Yamato K T,Kohchi T,et al. n-Hexanal and(Z)-3-hexenal are generated from arachidonic acid and linolenic acid by a lipoxygenase inL.[J]. Bioscience Biotechnology & Biochemistry,2017,81(6):1148-1155.
[7] Takemura M,Maoka T,Misawa N. Biosynthetic routes of hydroxylated carotenoids(xanthophylls)in,and production of novel and rare xanthophylls through pathway engineering in. [J]. Planta,2015,241(3):699-710.
[8] Akashi H,Okamura E,Nishihama R,et al. Identification and biochemical characterization of the serine biosynthetic enzyme 3-phosphoglycerate dehydrogenase in[J]. Frontiers in Plant Science,2018,9:956.
[9] Tsuboyama-Tanaka S,Kodama Y. AgarTrap-mediated genetic transformation using intact gemmae/gemmalings of the liverwortL[J]. Journal of Plant Research,2015,128(2):337.
[10] Tanaka D,Ishizaki K,Kohchi T,et al. Cryopreservation of gemmae from the liverwortL.[J]. Plant & Cell Physiology,2016,57(2):300-306.
[11] Saintmarcoux D,Proust H,Dolan L,et al. Identification of reference genes for real-time quantitative PCR experiments in the liverwort[J]. Plos One,2015,10(3):e0118678.
[12] 孙红卫,吕春燕,祁爱琴,等. 综合评价中数据标准化的原理研究[J]. 中国卫生统计,2015,32(2):342-344.
Sun Hongwei,Lü Chunyan,Qi Aiqin,et al. Theoretical study on data standardization in comprehensive evaluation[J]. Chinese Health Statistics,2015,32(2):342-344(in Chinese).
Constructing a Simple and Efficient System for Analysis of Gene Expression in
Miao Zhiqi,Zhou Lu,Xu Meihui
(School of Agriculture and Biology,Shanghai Jiao Tong University,Shanghai 200240,China)
Real time fluorescence quantitative polymerase chain reaction (qRT-PCR) has recently become widely used in the differential analysis of gene transcription. Stable expression of the reference genes, ideal reference primers designed to amplify the reference genes, and standardized treatment samples are the necessary preconditions for reliable qRT-PCR. With the advantages of having a small genome, short growth cycles, easy cultivation and reproduction, and high efficiency in transgenesis,is one of the new model plants used in synthetic biology. A large number of transgenic and mutant lines have been produced for the study of plant synthetic biology. Meanwhile, improvements to efficiency and reliability of qRT-PCR onhave been proposed through improvements to the speed and accuracy of analyzing the exogenous and endogenous gene expression levels. Bioinformatics analysis of thegenome showed that the reference actin gene ACT7 had double highly homologous gene sequences, and the expression of the two genes was complementary. However, the expression of either gene was not very stable. In order to construct a simple and efficient system to analyze gene expression in qRT-PCR for, a mathematical model was used to analyze the error conduction mechanism. A comprehensive evaluation index system for the reference primers was also introduced. We found that the co-annealing primer expression level was made more stable by amplifying the two highly homologous ACT7 genes. Primers numbered 3—6 showed overall superiority on multiple indicators,such as amplification efficiency, expression level, and stability. In this way, a qRT-PCR platform for rapid and accurate detection of gene expression level was developed inThis method can guide future research aiming to optimize the qRT-PCR detection systems in other species.
qRT-PCR;;ACTIN reference gene;co-annealing primers design
10.11784/tdxbz201808072
Q946.2
A
0493-2137(2019)06-0661-08
2018-08-24;
2018-10-19.
苗志奇(1974—),男,博士,副教授.
苗志奇,zqmiao@sjtu.edu.cn
国家自然科学基金资助项目(30700061);上海科技支撑计划资助项目(144319057001);国家重点基础研究发展计划(973计划)资助项目(2015CB755700).
the National Natural Science Foundation of China(No.30700061),the Shanghai Science and Technology Support Project (No.144319057001),the National Basic Research Program of China(No.2015CB755700).
(责任编辑:田 军)
