Proteomic Studies of Petal-specific Proteins in Soybean [Glycine Max (L.)Merr.] Florets
2019-04-12GuoFangliangLiuHanmiaoLuoTingtingFangSijiaPangZeYangMingmingWeiXiaoshuangSongBoLiuShanshanandLiWenbin
Guo Fang-liang, Liu Han-miao, Luo Ting-ting, Fang Si-jia, Pang Ze, Yang Ming-ming, Wei Xiao-shuang,Song Bo, Liu Shan-shan, and Li Wen-bin*
1 Key Laboratory of Soybean Biology of the Chinese Ministry of Education (Key Laboratory of Soybean Biology and Breeding/Genetics of the Chinese Agriculture Ministry), Northeast Agricultural University, Harbin 150030, China
2 College of Life Sciences, Heilongjiang University, Harbin 150080, China
Abstract: A survey of petal-specific proteomes of soybean (Glycine max (L.) Merr[Non-italic].) was conducted comparing protein expression profiles in different petals. Two-dimensional polyacrylamide gel electrophoresis reference maps of protein extracts from standard petals (SP), lateral wings (LW), keel petals (KP), and reproductive organs (RO) (a mixture of stamen and carpel) were obtained. Protein expression in the three petal types was compared using Image Master TM 2D platinum 6.0 software. This indicated that the proportion of homologous proteins between SP and LW was 59.27%, between SP and KP was 61.48%, and between LW and KP was 60.05%. Within a mass range of 6.5-200.0 ku and pH 4.0-7.0, approximately 590, 646, 544, and 700 protein spots were detected in SP, LW, KP, and RO, respectively. A total of 82 differentially expressed proteins were detected. Sixty-four of these detected spots were differentially expressed and showed more than 2-fold changes in abundance; of these 64 proteins, 26 showed increased expression and 38 showed decreased expression. Among these spots, single organ-specific proteins were also identified.They were ID 49 (60.9 ku), ID 45 (50.0 ku), and ID 46 (40.5 ku) in RO, ID 98 (42.0 ku) in SP, and ID 05 (29.0 ku) in KP. A total of 14 protein spots from 82 differentially expressed proteins were identified with LC-MS/MS. Further protein identification was conducted using the SwissProt and NCBInr databases. The identified proteins and their putative functions were discussed further. This was thefirst study reporting the comparison of petal protein profiles of soybeanflorets using proteomics tools.
Key words: soybean (Glycine max (L.) Merr.), 2D-PAGE, LC-MS/MS, petal protein
Introduction
In recent years, the application of proteomics tools,such as two-dimensional (2-D) gel electrophoresis in combination with advanced mass spectrometry(MS), has become popular. These tools are powerful methodologies for accurately detecting and examining changes in protein composition. Matrix-assisted laser desorption ionization/time-of-flight-MS (MALDITOF-MS) and liquid chromatography-electrospray ionization tandem-MS (LC-ESI-MS) are the keys to high-fidelity, high-throughput proteomics, and the two techniques have become the preferred methods for ionization of peptides and proteins because of their effective application to a wide range of proteins and peptides (Fenn et al., 1989; Karas et al., 2015;Link et al., 1999; Liska et al., 2010; Nyman, 2002;Porubleva et al., 2015). Several soybean (Glycine max (L.) Merr.) proteomics studies have recently been published, with some focused on different tissues,such as the nodule cytosol (Oehrle et al., 2008), leaves(Xu et al., 2006), and seeds (Mooney et al., 2004;Natarajan et al., 2006), whereas others have focused on the comparative analysis of proteomics profiles of soybean genotypes (Zarkadas et al., 2007; Natarajan et al., 2007). However, limited studies are available for soybeanfloret proteomes.
Soybean flowers are structurally similar to those of beans (Phaseolus vulgaris), peas (Pisum sativum),and Lotus japonicus and have three types of petals that are named the standard petal, lateral wings, and keel petal. The molecular mechanism involved in thefloral patterning and floral development in Leguminosae(Rosid clade of eudicots) (Benlloch et al., 2003;Bowman et al., 1991; Handberg and Stougaard, 2010;Lohmann et al., 2001), which is the third largest family offlowering plants with approximately 20 000 species, has been extensively studied at the genomic level in the model legume L. japonicus (Tucker,2003; Dong et al., 2005) and pea (Pisum sativum)(Taylor et al., 2002). However, little is known about the proteomes of different soybean (Glycine max)petals. Proteomic studies of soybean petals will help to define the specific changes in protein levels and composition that can occur in different petal types of soybean florets; the combination of proteomics and genetic analysis will facilitate a better understanding of the molecular mechanism of soybeanfloral patterning.
The objective of the present work was to survey the petal-specific proteomes of soybean to provide an overview of the soybean corollas-proteome and to serve as a basis for future proteome comparison of genetic mutants and for the modification of soybeanfloral patterning. The survey was accomplished using 2-DE to produce reference maps of proteins extracted from SP, LW, KP, and RO. Liquid chromatographyelectrospray ionization and ion trap mass spectrometry were used to identify differentially expressed proteins.The Mascot search engine was used to search against the Swissprot and NCBInr databases to identify proteins. The results provided a basis for future studies offloral differentiation and the functional contributions of petal-specific proteins.
Materials and Methods
Plant materials
Soybean petals were sampled from a normal plant at Northeast Agriculture Soybean Breeding Center(Harbin, China) in 2008. Before sampling, 1.5 mL Eppendorf tubes were labeled SP, LW, KP, and RO,respectively, and werefilled with liquid nitrogen. SP,LW, and KP petals of newly opened flowers were removed with tweezers and immediately placed in the prepared 1.5 mL Eppendorf tubes to prevent protein degradation. Sample tubes were then closed (each tube had a hole drilled into the center of the cap to facilitate nitrogen evaporation) and stored at -80℃ for further analysis. Three biological replicate samples were used for protein extraction and 2D-PAGE analysis.
Protein extraction from soybean florets,modified TCA/acetone precipitation, and 2D-clean up kit extraction
Frozen samples were ground in a mortar with liquid nitrogen, incubated with 10% TCA and 0.07% 2-mercaptoethanol in acetone for 3 h at -20℃, and then centrifuged for 20 min at 13 000 r · min-1at 4℃. The precipitated proteins were pelleted and washed with ice-cold acetone containing 0.07%2-mercaptoethanol to remove pigments and other interfering substances until the supernatant was colorless. Then, the pellet was vacuum dried, and the acetone dry powder was resuspended in resolubilization solution (5 mol · L-1Urea, 2 mol · L-1Thiourea, 2% CHAPS, 2% SB3-10, 5 mmol · L-1TCEP, and 20 mmol · L-1DTT) and sonicated to extract proteins. Insoluble tissue was removed by centrifugation at 13 000 r · min-1for 30 min and the supernatant was used in the 2D-PAGE analysis. The protein concentration was determined according to the Bradford method (Bradford, 1976). Aliquots of supernatant containing 250 μg of protein were used for 2D-PAGE analysis.
2D-PAGE analysis
Thefirst dimension IEF was performed using 18 cm,pH 4.0-7.0 linear IPG strips in an IPGphor apparatus,according to the manufacturer's recommendations.A protein molecular weight standard was used in the second dimension in each gel. IPG strips were rehydrated for 16 h with 400 μL rehydration buffer(5 mol · L-1Urea, 2 mol · L-1Thiourea, 2% CHAPS,2% SB3-10, 5 mmol · L-1TCEP, 20 mmol · L-1DTT,and 0.05% (W/V) bromophenol blue) containing 250 μg protein. The voltage settings for IEF were 100 V for 2 h, 200 V for 2 h, 1 000 V for 4 h, and 1 000 V to a total of 10 000 V. Following electrophoresis, the protein in the strips was denatured with equilibration buffer (50 mmol · L-1Tris-HCl,pH 8.8, 6 mol · L-1urea, 30% glycerol, 2% SDS, 0.002%bromophenol blue, and 1% DTT) and then incubated with the same buffer containing 2.5% iodoacetamide instead of DTT for 30 min at room temperature. The second-dimension electrophoresis was performed on a 12.5% gel using an Ettan DALTsix electrophoresis unit (GE Healthcare). The gels were stained with CBB-250 and scanned using an Imagine Scanner ⅢLanScan 6.0. All the inductions and two-dimensional protein gel analyses were performed at least three times.
In-gel digestion of protein spots
Protein spots were excised from the stained gel and washed with CH3CN : H2O (1 : 1, v/v, 100 µL)containing 25 mmol · L-1ammonium bicarbonate to remove the dye. The gel plug was dehydrated with 100% acetonitrile, dried under a vacuum, and then incubated overnight at 37℃ with 20 µL of 12.5 µg · mL-1trypsin in 25 mmol · L-1ammonium bicarbonate. Resulting tryptic fragments were eluted by diffusion into CH3CN : H2O (1 : 1, v/v) and 0.5%trifluoroacetic acid. A sonic bath was used to facilitate the diffusion. The extract was vacuum dried and dissolved in 5 µL of 50% acetonitrile and 0.1% trifl uoroacetic acid.
LC-ESI-MS/MS analysis
Selected digest mixtures were analyzed by nanoscale HPLC coupled with LC/MS/MS. The instrument m/z was calibrated with standards supplied by the manufacturer. Separated peptides were introduced automatically into the mass spectrometer with an HPLC system. Separations were achieved using an LC Packing PepMap C18column (2.1×30 mm i.d.,3.5 μm). The trypsin-digested samples were centrifuged for 10 min at 13 000 r · min-1at 4℃ and then the supernatant wasfiltered through 0.4 µmfilters. Next,8 μL of each prepared sample was injected into an HPLC (Agilent 1 200, Nano-flow, USA) system. The HPLC was operated in the trap mode. Mobile phase A was 0.1% formic acid and B was 100% acetonitrile.As peptides were eluted into the mass spectrometer data-dependent acquisition was performed. In this experiment, the mass spectrometer was set to acquire a conventional scan over the m/z range 300 2 200.The most intense peak was selected for MS/MS experiments. Protein identification was performed using the Mascot search engine (Matrix Science,http://www.matrixcience.com) (Apweiler et al., 2002;Bateman et al., 2008; Perkins et al., 1999).
Results and Discussion
One-dimensional electrophoresis of total proteins from soybeanflorets
To identify differences in protein expression in different soybean petals, protein extracts were analyzed by SDS-PAGE. Characteristic electrophoretic protein profiles of the total protein obtained from mature florets (newly opened flower), young florets (1-mmlong buds), SP, LW, KP, and RO are illustrated in Fig. 1. In 1-DE gel, about 26 sharp bands of polypeptides ranging from 14.3-97.2 ku, estimated by Quantity One software (Bio-Rad), were clearly resolved with a low background over the entire gel.Patterns among different floret organs were generally similar, except for major variations in the band patterns ranging from 14.3 to 29.0 ku. Onedimensional SDS-PAGE data indicated that there was variation in the protein band patterns between mature and young florets and within different petals.The protein bands of soybean petals could be divided into three classes: (1) protein bands common to all of the extracts examined (Fig. 1, open arrows);(2) protein bands observed in two kinds of petal but not clearly represented in all types (Fig. 1, red arrows on Lanes 3 and 5); and (3) protein bands detected in only one petal (Fig. 1, blue arrows on Lanes 3 and 4).The identity of the major protein bands presented in soybeanfloret organs was confirmed in separate 2-DE reference maps and LC-ESI-MS/MS analysis.
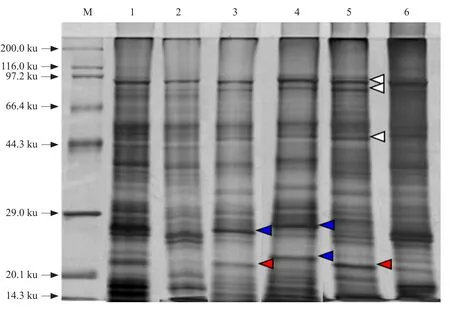
Fig. 1 One-dimensional SDS-PAGE of proteins extracted from various petal types of soybean corollas
2-DE separation and analysis
Initial 2-DE studies with IPG strips ranging from pH 3.0 to 10.0 showed that the proteome region from pH 4.0 to 7.0 was highly dense. For this reason, all the subsequent 2-DE studies (Fig. 2) were carried out using narrow-range IPG strips; the 2-DE reference maps shown in Fig. 2 were obtained using IPG strips that ranged from pH 4.0 to 7.0. Protein spots from different petals were compared with those from RO and between different petals. Two-dimensional electrophoresis gel image comparison using Image Master TM 2D platinum 6.0 software revealed that the number of protein spots and spot intensity of the extracted proteins varied among different petal types. SP, LW, and KP were found to share 53.16%,64.48%, and 59.68% of their polypeptides with that of RO, respectively, whereas the proportion of homologous protein between SP and LW was 59.27%, between SP and KP was 61.48%, and between LW and KP was 60.05%. Within a Mr of 6.5-200.0 ku, the total number of protein spots detected in SP, LW, KP, and RO was approximately 590, 646, 544, and 700, respectively. A total of 82 differentially expressed proteins were detected; 64 of these detected spots were found to be differentially expressed and showed more than 2-fold changes in abundance. Of these 64, 26 proteins showed increased expression and 38 showed decreased expression.
As shown in Fig. 2, among the 82 spots, several protein spots were identified by their presence or absence in other floret organs. Spot IDs 38, 40,41, 42, 47, 48, 35, 36, 31, 33, 21, and 22 represented in KP and RO, but were not observed in SP and LW. Spot ID 01 (22.0 ku) was restricted to extracts prepared from SP and KP, but was not detected in LW and RO. Single organ-specific proteins were also identified, including ID 49 (60.9 ku), ID 45 (50.0 ku),and ID 46 (40.5 ku) in RO, ID 98 (42.0 ku) in SP, and ID 05 (29.0 ku) in KP.

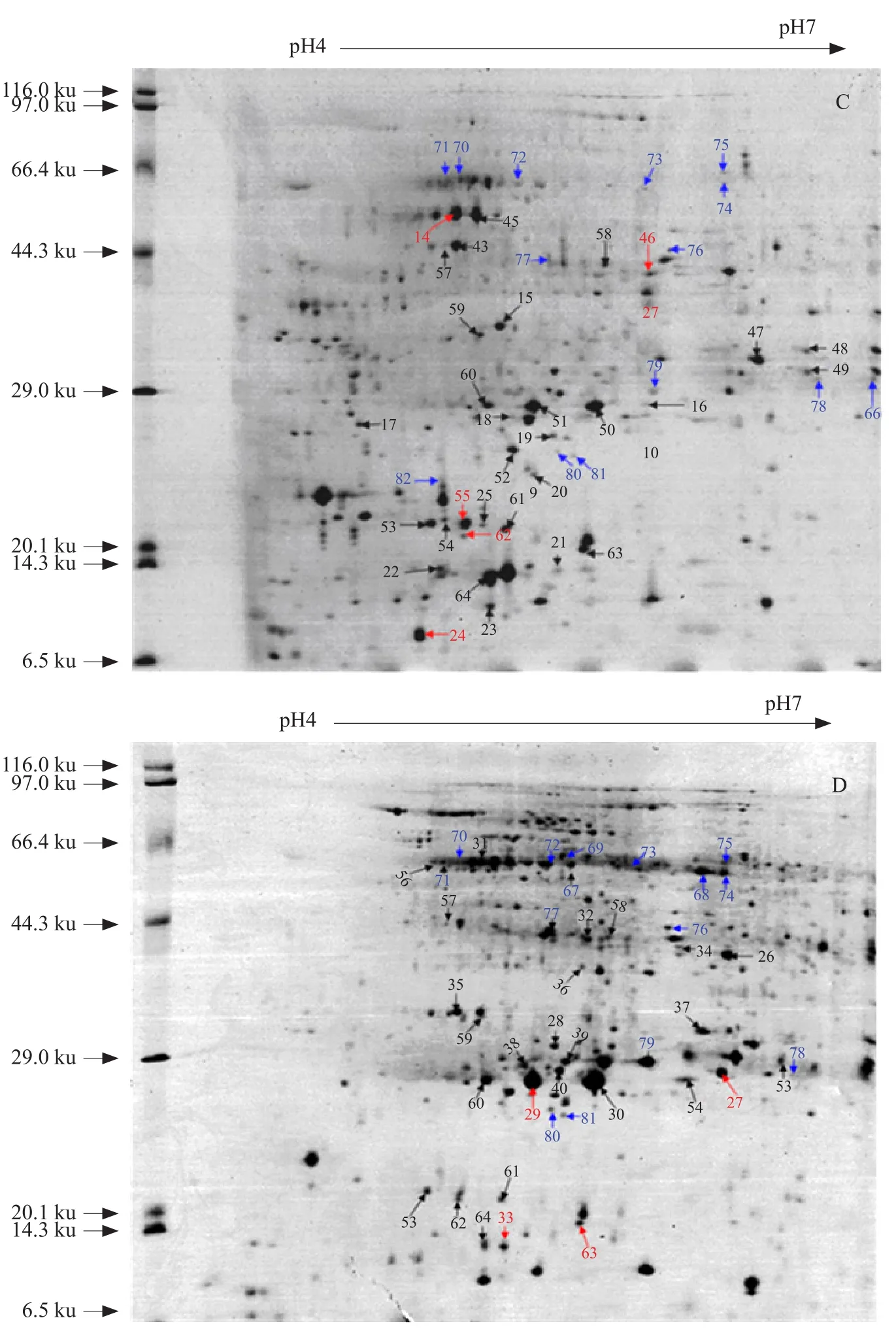
Fig. 2 Two-by-two comparison of 2-DE gel maps of proteins isolated from A. SP, B. LW, C. KP, and D. RO
ldentification of separated proteins
To qualitatively survey the proteins visualized by 2D-PAGE, a total of 64 protein spots were excised from the 2D-PAGE gels and digested with trypsin.The peptide fragments were extracted and analyzed by LC-MS/MS. Typically, high-quality LC-MS/MS peptide mass maps were obtained; of the 64 pro-tein spots extracted from the gel, only 14 proteins were confidently identified by querying the SwissProt and NCBI databases using the Mascot search engine.Many spectra matched only clones with no ascribed information (data not shown). Identities of protein spots from SP, LW, KP, and RO are listed in Table 1.

Table 1 Proteins identified from soybean petals by LC-MS/MS
Data in Table 1 included an assigned protein spot number (SID), protein identity and its original species,theoretical Mr and PI (T. Mr/pI), MOWSE score(MO), number of peptides matched (PM), percentage sequence coverage (SC), and the accession number(Acce. No) of the best match. Relative to other petals,a large percentage (i.e., 80%) of the soybean KP proteins were identified as actin. It was possible that actin might be specific to KP and might be involved infloral patterning of soybean corolla development.
Protein classification and assembly of a webbased database
The identified proteins were classified based on putative functions determined through similarity (Bevan et al., 1998). Summaries of protein functions observed in the soybean floret proteome are provided in Fig.3. Some proteins could be unambiguously classified while other proteins were associated with multiple functions; classification of these proteins was based on their predominate functions. The "unclear" protein class included proteins that were successfully matched to putative proteins from such sources as the Pinus halepensis genomic sequence, but did not yet have a known function. The identified proteins were categorized into five groups by function (% of total protein analyzed): cell structure proteins (43%);disease/defense proteins (29%); protein destination and storage (14%), metabolism (7%), and proteins of unclear classification (7%). Discussions concerning a portion of the proteins observed and their functional role were presented below in relation to the petal in which they were observed.
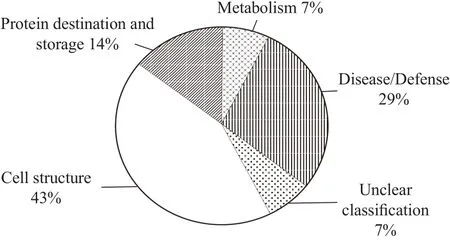
Fig. 3 Assignment of identified proteins in functional categories using classification described by Bevant
Several proteins identified in SP and LW were related to disease/defense or were involved in carbon metabolism. One of these was a dehydroascorbate reductase (spot 27) detected in SP. The expression of dehydroascorbate reductase, responsible for regenerating ascorbic acid from an oxidized state,regulated the ascorbic acid cellular redox state that in turn affects cell responsiveness and tolerance to environmental reactive oxygen species. The disease/defense protein identified in LW (spot 66) was a trypsin inhibitor. A trypsin inhibitor was a potent inhibitor of trypsin and might have a protective function against plant pests and pathogens. One enzyme responsible for carbon metabolism, malate dehydrogenase (spot 24), was also identified in SP. As mentioned above, these enzymes were commonly associated withflower pigmentation or ascorbic acid recycling and serve as important defense proteins against insect and pathogen attacked as well as possible antinutritional factors. One of the main functions of the corolla was enclosing and protecting the stamen and pistil. Therefore, it was not surprising that differentially expressed proteins in the corolla proteome were associated with the disease/defense category.
Interestingly, this comparative proteomics analysis indicated that actin was highly represented in soybean KP. In the study, approximately 43% of the identified protein spots were associated with cell structure and included actin and keratin (typeⅡcytoskeletal). Plant actin was one of the important components of the cytoskeleton; it was necessary for the growth, differentiation, and reproduction of plant cells and participates in many important cellular processes, including muscle contraction, cell motility,cell division and cytokinesis, vesicle and organelle movement, cell signaling, and the establishment and maintenance of cell junctions and cell shapes.The fact that actin was one of the most abundant differentially expressed KP proteins suggested that this protein might perform a critical role in KP function.Conversely, actin protein was also differentially expressed in other petal types such as SP.
Proteins involved in the disease/defense category were also detected in RO. They were dehydroascorbate reductase (spot 123) and lipoxygenase (spot 114).Other identified RO protein spots were included in the protein destination and storage category, such as vegetative storage protein 1 (spots 125 and 126).
Conclusions
Soybean floret proteins were separated using 2DPAGE and 23 protein spots were identified with LCMS/MS. The results from the current study indicated that the natural variation of polypeptides occurred in different soybean petal types. One-dimensional SDSPAGE data indicated that there was variation in the protein band patterns between mature florets (newly opened flowers), young florets (1-mm-long buds),and within different petal types. Conversely, two-bytwo comparison of 2-DE gel maps indicated petalspecific proteins, including a 42.0 ku (spot 65) protein presented in SP and a 29.0 ku (spot 66) protein presented in KP. Actin was the most highly represented petal-specific protein in KP and was involved in the cell structure. The identified proteins were categorized into five groups by function (% of total protein analyzed); cell structure proteins (39%),disease/defense proteins (17%), protein destination and storage (9%), metabolism (9%), and proteins of unclear classification (26%). Future studies of floral patterning differentiation and corolla development in soybean would benefit from this proteome reference map of soybean petals.
Acknowledgments
Liu Shan-shan and Li Wen-bin contributed equally to this article.
杂志排行

Journal of Northeast Agricultural University(English Edition)的其它文章
- Effect of GmLEC1-A Expression on ABA Content at Germination Stage in Soybean (Glycine max)
- Compatibility Screening of Plant Extracts Synergistic with Osthole
- Bioinformatics and Expression Pattern Analysis of Tomato nsLTP 2-like cDNA full-length Gene Clone
- Molecular Differentiation of Different Pathogenic Phenotypes of Infectious Bursal Disease Viruses by RT-PCR Combined with Restriction Fragment Length Polymorphism (RFLP) Assay
- Effect of Xylazine Anesthesia on Glu and GABA Amino Acid Neurotransmitters in Rat
- Chlorophyll Content Retrieval of Rice Canopy with Multi-spectral Inversion Based on LS-SVR Algorithm