气相色谱串接质谱测定食醋中多组分农药残留含量的方法研究
2019-04-09卞晶晶
李 丽,夏 蓉*,卞晶晶
(1.江苏联合职业技术学院 镇江分院镇江高等职业技术学校 机电工程系,江苏 镇江 212000;2.镇江恒顺生物工程有限公司,江苏 镇江 212021)
镇江醋素有“天下美酒推茅台,世上食醋数镇江”之说,其原料主要包括大米(糯米、粳米、籼米)、辣蓼草、麸皮、大麦、小麦、豌豆、黄酒糟、食盐、食糖等。镇江醋因其“酸而不涩,香而微甜,色浓味鲜,愈存愈香”的特色,受到国内外消费者的青睐。2006年6月,经历了170多年有序传承的“镇江恒顺醋酿制技艺”被列入第一批国家非物质文化遗产名录。2012年7月,中国镇江醋成为首个在欧盟注册的食醋类地理标志保护(protected geographic indication,PGI)产品。欧盟、美国、日本等国也成为镇江食醋的主要出口目的地[1]。
对于植物源性的产品(如糯米、小麦等),欧盟[2]、美国[3]、日本都有明确的农药残留限量的规定。近年来,这些国家实施了越来越严格的残留限量的要求,其中欧盟对于未制定最高残留限量的农药实施了“一律标准”,均不得检出(检出限10 μg/kg)[4],这对中国农产品的出口带来考验,对以粮食为主要原料酿制的食醋同样有着严重的影响[5]。因此,在严格控制生产原料质量、确保相关农药残留合格的同时,对最终产品中农药残留的检测也尤为重要。目前,植物源性产品中农药残留的检测方法包括液相色谱质谱(liquid chromatography-mass spectrometry,LC-MS)法[6-9]、气相色谱质谱(gaschromatography-mass spectrometry,GC-MS)法[10]、气相色谱(gas chromatography,GC)法[11-12]等。提取待测组分经常采用的试剂包括乙腈、乙酸乙酯、甲苯、丙酮、二氯甲烷等。但这些提取、测定方法主要针对蔬菜、水果、谷物以及一些动物源性类的产品,没有专门针对醋中农药残留提取的描述,缺乏针对性的检测方法。
因此,本研究选择了有机氮、甲氧基丙烯酸酯、拟除虫菊酯、三唑类、喹唑啉类、杂环类、有机磷、二甲酰亚胺类、醚类、吡咯类、氨基甲酸酯类、酰胺类、苯基吡唑类、嘧啶胺类、苯胺类、芳香族化合物、取代硝基苯、联苯肼等共计19类农药进行样品前处理的研究,在此基础上建立了食醋中多组分农药残留的气相色谱-串联质谱(gas chromatographytandem mass spectrometry,GC-MS/MS)的检测方法,并对市售食醋产品进行筛查,积累农药残留的本底数据,确保食醋的质量安全。
1 材料与方法
1.1 材料与试剂
50份镇江食醋:当地超市。
乙腈、环己烷、甲苯、丙酮、二氯甲烷(均为色谱纯):德国Merck公司;氨水、氯化钠(均为分析纯):上海国药集团;N-丙基乙二胺(primarysecondaryamine,PSA)固体粉末:上海安普公司;石墨化炭黑(graphitized carbon blacks,GCB):深圳逗点公司;农药标准品(纯度均>98.5%):德国Dr.Ehrenstorfe公司。
1.2 仪器与设备
Agilent 7890A气相色谱仪、Agilent 7000B GC-MS/MS仪、Agilent 7693自动进样器:美国安捷伦公司;ME-T电子天平、SevenExcellence pH计:瑞士梅特勒公司;3K15高速冷冻离心机:德国Sigma公司;XW-80A涡旋混合器:上海医科大学仪器厂。
1.3 方法
1.3.1 样品前处理
本研究采用乙腈、甲苯、丙酮、二氯甲烷、环己烷溶剂对醋中的农药进行提取,考察不同萃取剂对待测组分的提取效率和样品基质的干扰。以样品基质干扰小、回收率符合相关的技术要求为依据,优化组合后[13-14],确定样品的提取和净化程序:量取100 mL食醋于200 mL烧杯中,在pH计上逐滴加入氨水中和至pH 7.0。吸取3.0 mL中和后的食醋液于50 mL离心管中,加入2.0 g氯化钠,涡旋30 s。加入最优萃取溶剂,涡旋混匀3min。8000r/min离心5min后,有机相和水相结合处会有絮状物质,分层不清。用手轻轻摇晃离心管,让有机相和水相结合处的絮状物质彻底分散,再以13000r/min离心10min,取上清液10 mL净化。
净化:先用5 mL甲苯溶液清洗GCB小柱并保持柱床的湿润,取上清液10 mL过柱,过柱结束后用2 mL甲苯清洗GCB小柱,合并滤液,氮吹至干。残渣中加入1 mL乙腈和100 mg PSA,涡旋混匀2 min,过有机相膜,供GC-MS/MS测定。
1.3.2 检测条件
气相色谱条件:DB-5MS毛细管柱(30 m×0.25 mm×0.25 μm);载气为氦气(He);流速为1.0 mL/min;进样量为1.1 μL,不分流进样;进样口温度为250℃;程序升温条件:初始温度70℃,保持3 min;以10℃/min升至105℃,保持2 min;以10℃/min升至180℃,保持2 min;以6℃/min升至250℃,保持4 min;以5℃/min升至280℃,保持2 min;以20℃/min升至300℃,保持5 min。溶剂延迟10 min。
质谱条件:离子化模式为电子电离(electronionization,EI)源;电离能量为70 eV;离子源温度为230℃;GC-MS/MS接口温度为270℃;多反应监测(multiple reaction monitoring,MRM)模式。
定性定量:以待测组分的保留时间和离子对作为定性依据;以相对丰度较高的离子作为定量离子。
1.3.3 标准曲线的建立
吸取100.0μL52种农药的混合标准溶液(1250.0μg/L)于900.0μL乙腈溶液中,涡旋混匀。再用乙腈进行倍比稀释,使混合标准溶液的质量浓度分别为7.81 μg/L、15.60 μg/L、31.20 μg/L、62.50 μg/L、125.00 μg/L。以峰面积(Y)对质量浓度(X)绘制标准工作曲线。
1.3.4 方法回收率及精密度
用经过筛查的不含待测组分的空白样品进行加标回收试验,添加水平为10.0 μg/L、20.0 μg/L和40.0 μg/L,每个加标水平测定6次,得出方法的回收率及精密度。
1.3.5 方法检出限及定量限
参照文献[15]的方法,量取100 mL食醋于200 mL烧杯中,加入农药混合标准溶液290μL(1000.0μg/L),按照1.3.1进行样品的前处理。重复测定7次,以3∶1的信噪比确定方法的检出限(limit of detection,LOD),以10倍的信噪比确定方法的定量限(limit of quantitation,LOQ)。
1.3.6 食醋样品的检测
按照方法1.3.1对食醋中的残留农药进行提取和净化,采用气相色谱-串联质谱法进行检测。
2 结果与分析
2.1 样品前处理条件的选择
食醋中含有约5%的乙酸,乙酸能与乙腈互溶,在甲苯中也有一定的溶解度。因此,采用宋碧君等[16-17]的方法(盐析法)直接用乙腈、甲苯提取食醋中的农药残留,乙酸会将样品中的非目标化合物带入提取液中,基质干扰大。采用丙酮、二氯甲烷提取出的色素含量较多,不利于后续的样品提取液的净化。用环己烷提取,有机磷类的农药回收率偏低。以乙腈提取为例,空白样品中邻苯基苯酚、氧化乐果、克百威、硫环磷、苯醚甲环唑都呈假阳性,双甲脒的回收率<10%。
乙酸铵在乙腈、甲苯、环己烷等有机溶剂中溶解度都非常小甚至不溶解,因此将醋中的乙酸用氨水中和成乙酸胺,再用有机试剂提取,可以有效减少样品基质的干扰。因此,分别考察了乙腈、甲苯、环己烷的提取效果。单独用乙腈提取,硫环磷和双甲脒的回收率<10%,并且提取液中色素含量较高;纯甲苯提取,包括甲苯氟磺胺、喹螨醚、嘧菌酯等19种物质的回收率偏高;纯环己烷对于有机磷类农药的提取效果不好,回收率偏低。
参考文献[18]依据不同农药在甲苯和环己烷中的溶解度不同,采用甲苯和环己烷的混合溶液进行提取。甲苯/环己烷提取液中含有少量的色素,用GCB[19-20]净化可以有效去除这部分色素,减少样品提取液对色谱柱和质谱的污染,降低基质的干扰;PSA的使用可以有效降低样品基质对菊酯类物质的增益影响。因此,对甲苯、环己烷、PSA的用量进行了3因素3水平的正交试验[13-14],以样品基质干扰小、回收率符合相关的技术要求为判定依据,确定了甲苯/环己烷的提取方式以及样品提取液的净化方法,确定甲苯添加量为10 mL,环己烷添加量为5 mL,PSA添加量为50 mg。
2.2 52种农药标准品的保留时间和质谱参数
食醋中各待测组分的保留时间(retention time,RT)、特征离子、碰撞能量等参数见表1,52种农药混标(62.5 μg/L)的总离子流色谱图见图1。

表1 52种农药的保留时间、特征离子、碰撞能量等参数Table1 Retention time,characteristic ions and collision energy of 52 kinds of pesticides

续表

图1 多组分农药残留的GC-MS/MS总离子流图Fig.1 Total ion chromatograms of multi-component pesticides residue analysis by GC-MS/MS
由表1可知,各种农药均能产生丰富的碎片离子,用于定量、定性分析。由图1可知,52种农药组分在15.93~37.46min内分离效果良好,避免了各种农药之间相同碎片离子的相互干扰。
2.3 52种农药的标准曲线及线性范围
52种农药的标准曲线、线性范围见表2。

表2 52种农药的标准曲线、线性范围Table2 Standard curves and linearity ranges of 52 kinds of pesticides

续表
由表2可知,52种农药在质量浓度为7.81~125 μg/L的范围内,相关系数R2均>0.99,线性关系良好,可用于52种农药的检测。
2.4 精密度试验
食醋中52种农药的相对标准偏差(relative standard deviation,RSD)结果见表3。
由表3可知,食醋中52种农药的相对标准偏差(RSD)在5.90%~11.4%范围内,表明该方法精密度良好[21]。

表3 方法精密度试验结果Table3 Results of precision tests for the method
2.5 加标回收率试验
食醋中52种农药的加标回收率试验结果见表4。
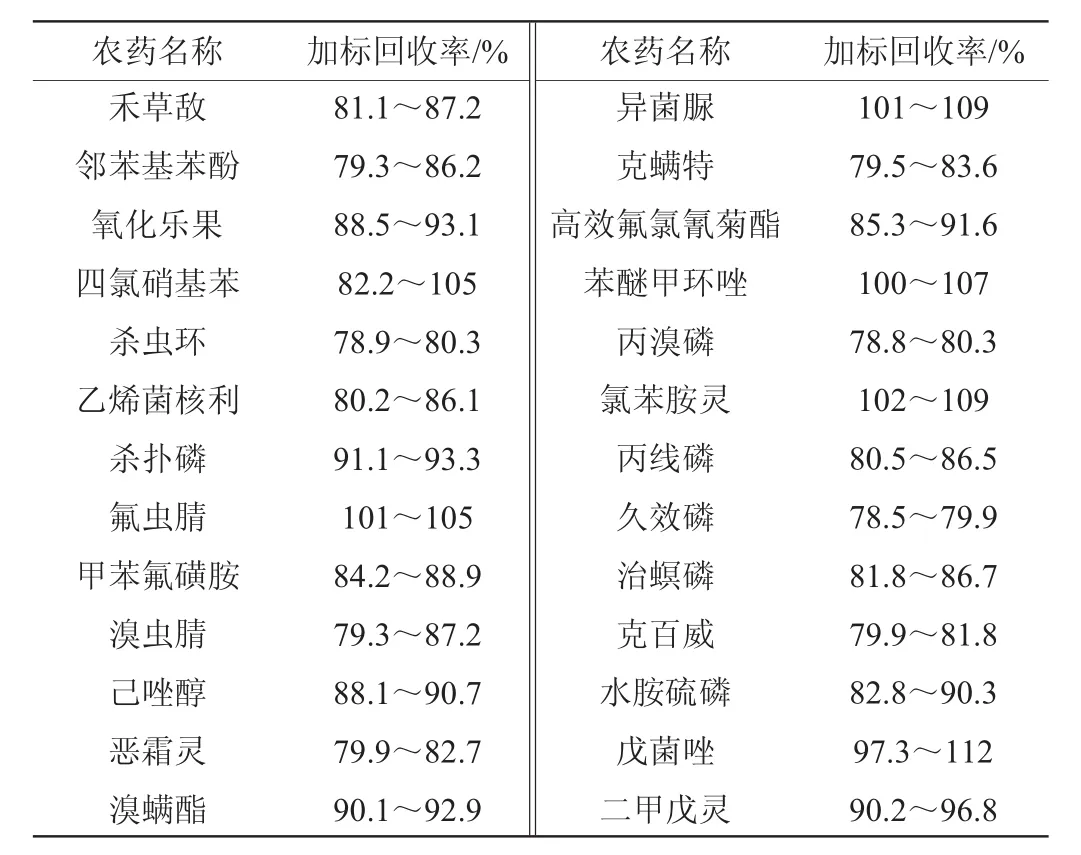
表4 加标回收率试验结果Table4 Results of adding standard recovery tests

续表
由表4可知,52种农药的回收率在78.5%~112%范围内,表明该方法的准确性良好[21]。
2.6 52种农药的检出限和定量限
52种农药的检出限均为5 μg/L,定量限均为10 μg/L。
2.7 食醋样品的检测
50份食醋样品中均未检出农药残留。
3 结论
在优化样品前处理条件以及仪器参数的基础上建立了GC-MS/MS法检测食醋中的多组分农药残留的测定方法。食醋经氨水中和后,采用甲苯/环己烷的混合溶液提取食醋中的待测组分,能有效降低醋的基质干扰。目标化合物在一定质量浓度范围内(7.81~125 μg/L)线性关系良好,相关系数R2均>0.99。方法的定量限和检出限分别为10.0 μg/L、5.00 μg/L,平均回收率为78.5%~112.0%,相对标准偏差(RSD)为5.90%~11.40%。该方法简便、快速,准确,已成功应用于市售食醋中农药残留的检测。为确保食醋的安全质量提供技术支撑。
