CCR3特异性拮抗剂GW766994在阿尔茨海默病发病机制中的作用研究
2019-04-03董春瑶
隋 轶,徐 冰,任 莉,董春瑶,张 尧
0 引言
阿尔茨海默病(AD)是目前老年人痴呆的主要原因之一,其主要病理标志包括由星形胶质细胞和微胶质细胞包围的细胞外淀粉样蛋白β(Aβ)沉积共同构成的老年斑;小胶质细胞、神经元纤维缠结的细胞内积累(由过度磷酸化的tau组成),以及神经元和突触的丧失[1-2]。绝大多数AD病例发病晚,通过治疗或预防能明显延迟发病年龄[3]。越来越多的证据表明,由活化的星形胶质细胞和小胶质细胞产生的促炎性细胞因子、趋化因子和神经毒素在AD的发病机制中起着重要作用[4]。由于它们在调节神经元-小胶质细胞交互影响中的关键作用,趋化因子在AD研究中引起了越来越多的关注[5]。因此,探讨趋化因子在AD病理学中的作用,有助于对AD发病机制的理解及开发新的AD治疗方法。
CCL11可能是参与AD病理学的一种重要趋化因子。在衰老过程中,CCL11在血清中逐渐升高,其年龄依赖性增加与海马依赖性学习和记忆任务的缺陷相关[6]。因此,CCL11很可能是影响海马功能的关键因素,并与AD有密切关联。相关机制可能是CCL11与一种由星形胶质细胞、小胶质细胞和神经元表达的趋化因子受体CCR3相结合[7-10]。本研究观察特异性CCR3拮抗剂GW766994是否通过拮抗趋化因子CCL11的作用影响AD的病理变化。
1 资料和方法
1.1 体视学细胞计数分析 应用荧光或光学显微镜进行体视学分析,油浸100×物镜。使用无偏立体细胞计数技术的光学分离器方法计数海马中Aβ斑和p-Tau阳性神经元的数量[11-12]。
1.2 蛋白质印迹分析[11]将细胞或脑组织在含有蛋白酶抑制剂混合物的TBS(20 mmol/L Tris-HCl 缓冲液,pH=7.4,150 mmol/L NaCl)中匀浆,包括0.5 mmol/L苯甲基磺酰氟,20 μg/ml 抑肽酶,20 μg/ml亮抑酶肽,20 μg/ml 胃蛋白酶抑制剂和1 ml EDTA(所有从Sigma获得的抑制剂)。将匀浆物短暂超声处理并以15 000×g离心30 min。在还原条件下,将样品(每泳道10 μg 蛋白质)在10%SDS聚丙烯酰胺凝胶上电泳。将蛋白质转移到聚偏二氟乙烯(GE Healthcare)膜上。用含有0.1%Tween 20、3%干乳的TBS封闭膜1 h,然后在室温下用特异性抗体孵育2 h。洗涤后,将印迹与相应的HRP标记的第二抗体(1∶1 000稀释)一起温育1 h。使用ECL系统(GE Healthcare)检测标记。使用光密度测定软件(Scion Image)分析条带。后续抗体用于蛋白质印迹:兔多克隆抗APPC末端片段(CTF)(Sigma),小鼠单克隆抗p-tau(T181)(1∶500;Abcam),兔多克隆抗p-tau(T205)(1∶1 000;Santa Cruz Biotechnology),兔多克隆抗p-tau(Ser199)(1∶1 000;Santa Cruz Biotechnology),兔多克隆抗p-tau(Ser235)(1∶1 000;Santa Cruz)Biotechnology),小鼠单克隆抗细胞周期蛋白依赖性激酶5(CDK5)(1∶1 000;Santa Cruz Biotechnology),小鼠单克隆抗p-ERK1/2(1∶1 000;Santa Cruz Biotechnology),小鼠单克隆抗p-CDK5(Ser159)(1∶200;Santa Cruz Biotechnology),小鼠单克隆抗p-糖原合成酶激酶3β(GSK3β)(Ser9)(1∶1 000;Santa Cruz Biotechnology),小鼠单克隆抗磷酸化GSK3α/β(Tyr279/216)(1∶1 000;ECM Biosciences),兔多克隆抗tau(1∶1 000;Santa Cruz Biotechnology),小鼠单克隆抗GSK3β(1∶4 000;Sigma),小鼠单克隆抗ERK2(1∶1 000;Santa Cruz Biotechnology),小鼠单克隆抗β-actin(1∶4 000;Sigma)。GW766994为同济大学范国煌教授馈赠。
1.3 树突棘的数量测定 利用Matlab 软件分析树突长度和这些神经元中蘑菇刺的数量。
1.4 原代神经元培养和转染[13]在胚胎第18~19天,从大鼠胎儿制备。在手术显微镜下解剖胚胎大鼠海马组织,并用0.15%胰蛋白酶在37 ℃消化10~15 min。通过添加10%补充的DMEM结束消化胎牛血清。用70 μm细胞过滤器过滤海马神经元悬浮液,然后在有或没有聚-D-赖氨酸氢溴酸盐包被的盖玻片的培养板中铺板。将细胞维持在补充有10%FBS和1%Pen/Strep的DMEM中。接种后12 h,用含有2% B27补充物、1% Glutamax和1%Pen/Strep的Neurobasal培养基替换培养基。对于形态学研究,使用Lipofectamine 2000在体外培养7 d瞬时转染EGFP载体的神经元培养物。在盖玻片上生长的神经元(14 DIV)用PBS洗涤,并在室温下在4%甲醛(pH 7.4)中固定15 min。然后用SlowFade Light试剂(Molecular Probes,Eugene,OR)安装在载玻片上。
1.5 原代海马神经元培养物中Aβ水平的ELISA分析 使用高度敏感夹心ELISA试剂盒(Immuno-Biological Laboratories,Gunma,Japan)测量原代细胞培养基中Aβ的浓度。简言之,单克隆兔抗人IgG抗Aβ35-40或抗Aβ38-42分别用于标记Aβ1-40和Aβ1-42。辣根过氧化物酶缀合的抗人Aβ11-28Fab用于以上2种测定中的检测[14]。该检测抗体识别Aβ1-40的整合和N-末端切割的变体和Aβ1-42。Aβ1-40和Aβ1-42的测量范围分别为4~231 pmol/L和3~178 pmol/L。
1.6 免疫细胞化学和共聚焦显微镜[15]将细胞用含有4%多聚甲醛的PBS(pH 7.4)固定15 min,然后用PBS中的0.5% Triton X-100膜透化5 min。在37 ℃下用3%牛血清白蛋白封闭1 h后,将细胞与以下一抗孵育:兔多克隆抗MAP2抗体(1∶200,Sigma)和小鼠单克隆抗CCR3抗体(1∶200,Santa Cruz),4 ℃过夜。然后将细胞与缀合至花青3(Cy3,1∶500;Jackson ImmunoResearch)或Alexa 488(1∶500;Molecular Probes)的二抗一起温育。使用具有25×或63×水浸物镜 LSM 510 Zeiss显微镜获得荧光图像。所有图像均以512×512像素分辨率拍摄。
2 结果
2.1 CCR3在14 DIV海马神经元中的表达 用抗CCR3和抗MAP2抗体共同免疫染色海马神经元培养物(14 DIV),发现CCR3和MAP2表达神经元之间的共定位(图1A),而用同种型对照IgG的免疫染色未显示CCR3信号(图1B),表明CCR3在神经元中的特异性表达。通过计数每组200个神经元,观察到100%神经元表达CCR3。

图1 CCR3在14 DIV海马神经元中的表达
注:免疫荧光显示海马神经元培养物中MAP2的CCR3的共定位(14 DIV)。Scale bar=50 μm
2.2 CCL11处理导致CDK5的激活 CCL11在治疗野生鼠(非CCR3- / -小鼠)的海马神经元时(图2),可导致CDK5的激活,CDK5是一种蛋白激酶,在AD发病机制中起重要作用[16]。

图2 CCL11处理导致CDK5的激活
注:来自野生型或CCR3- / -小鼠的海马神经元培养物(14 DIV)的细胞裂解物中,在CCL11(10 nmol/L)不存在或存在条件下共培养30 min,磷酸化CDK5(Ser159)和CDK5的蛋白质印迹试验结果
此外,CCL11诱导的CDK5活化被CCR3特异性拮抗剂GW766994(10 μmol/L),以剂量依赖的方式所逆转(图3)。

图3CCL11诱导的CDK5活化被CCR3特异性拮抗剂剂量依赖性逆转
注:A.在存在或不存在不同浓度GW766994的情况下,给予或不给予CCL11(10 nmol/L)处理的30 min海马神经元培养物(14 DIV)细胞裂解物中磷酸化CDK5(Ser159)和CDK5的蛋白质印迹试验结果。B.磷酸化CDK5的定量分析。数据表示为未处理样本值的倍数。通过单向ANOVA分析,与未处理组相比,*P≤0.05,**P≤0.01
2.3 CCL11处理导致CDK5和GSK 3β磷酸化的时间依赖性增加,从而介导Tau蛋白磷酸化增加 处理海马神经元培养物(14 DIV)与CCL11(10 nmol/L)不同的时间间隔后,导致CDK5和GSK3β磷酸化时间依赖性增加,后者在AD发病机制中也起着重要的作用[16]。CDK5和GSK3β磷酸化可以通过GW766994(10 μmol/L)(一个CCR3特异性拮抗剂)预处理而逆转,表明这些信号传导途径是通过CCR3介导的(图4)。
主要通过CDK5和GSK3β介导的Tau蛋白磷酸化是AD的主要标志,用CCL11(10 nmol/L)处理海马神经元(14 DIV)15 min,导致在几个测试位点(包括T181、T205、S235、S199)的tau磷酸化增加,并且这种变化可被GW766994(10 μmol/L)所逆转(图5、图6)。
2.4 CCL11处理导致神经元中Aβ产生的时间依赖性增加 从体外第14天开始,用对照或不同浓度的CCL11处理原代海马神经元细胞48 h,CCL11处理的细胞中观察到显著的时间依赖性Aβ1-42分泌增加,此变化同样可被GW766994(10 μmol/L)所逆转。针对CCL11的处理,Aβ 1-40水平也出现了增加,但这种影响并没有达到统计学意义(图6B)。为了补偿培养基的任何不均匀蒸发,对培养基样品进行Bradford蛋白质测定,数据以pmol Aβ/100μg蛋白质表示(图6)。
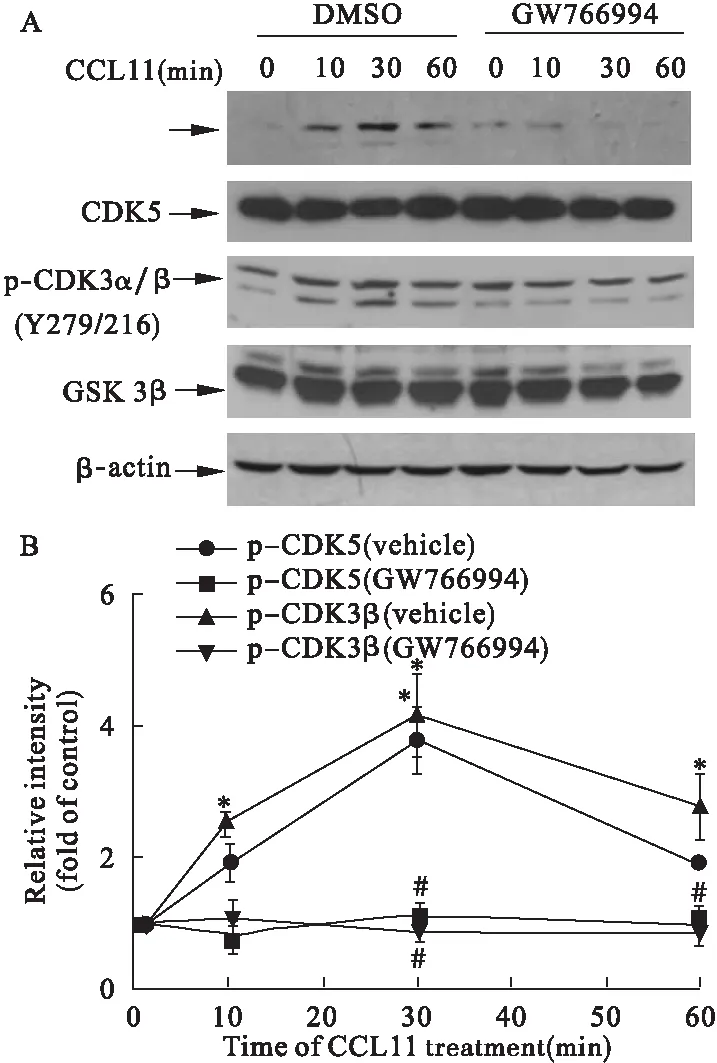
图4 CCL11处理导致CDK5和GSK3β磷酸化的时间依赖性
注:A.在海马神经元培养物(体外14 d)的细胞裂解物中给予CCL11(10 nmol/L)处理,同时加用或不加用GW766994(10 μmol/L),不同时间间隔条件下的磷酸化CDK5(Ser159)、CDK5、磷酸化GSK3α/β(Tyr216)和GSK3β结果。B.磷酸化的CDK5、GSK3β的定量分析结果,结果分别表示为未经处理的细胞值的倍数。数据来自3个独立实验。通过单向ANOVA分析差异。与同一治疗组的零时间点比较,*P<0.05,**P<0.01;与用载体处理的细胞相比,#P<0.05,##P<0.01
2.5 CCL11处理导致神经元形态改变 表达EGFP的海马神经元(14 DIV)在有/无GW766994(10 μmol/L)的条件下用/不用CCL11(10 nmol/L)处理,对树突状复杂性和树突棘密度进行评估。结果显示,CCL11处理导致树突状交叉(图7)数量及树突棘(图7)密度显著减少,此变化可以被GW766994(图1)的预处理所逆转。
3 讨论
衰老期间趋化因子在血清中水平升高,目前,其在神经变性疾病中的作用正在引起越来越多的关注。在本研究中,我们证明了诱导CCL11介导了海马神经元培养物中CDK5和GSK3β的活化,与升高的tau磷酸化相关联,增强Aβ的产生,并加速树突棘的缺失。
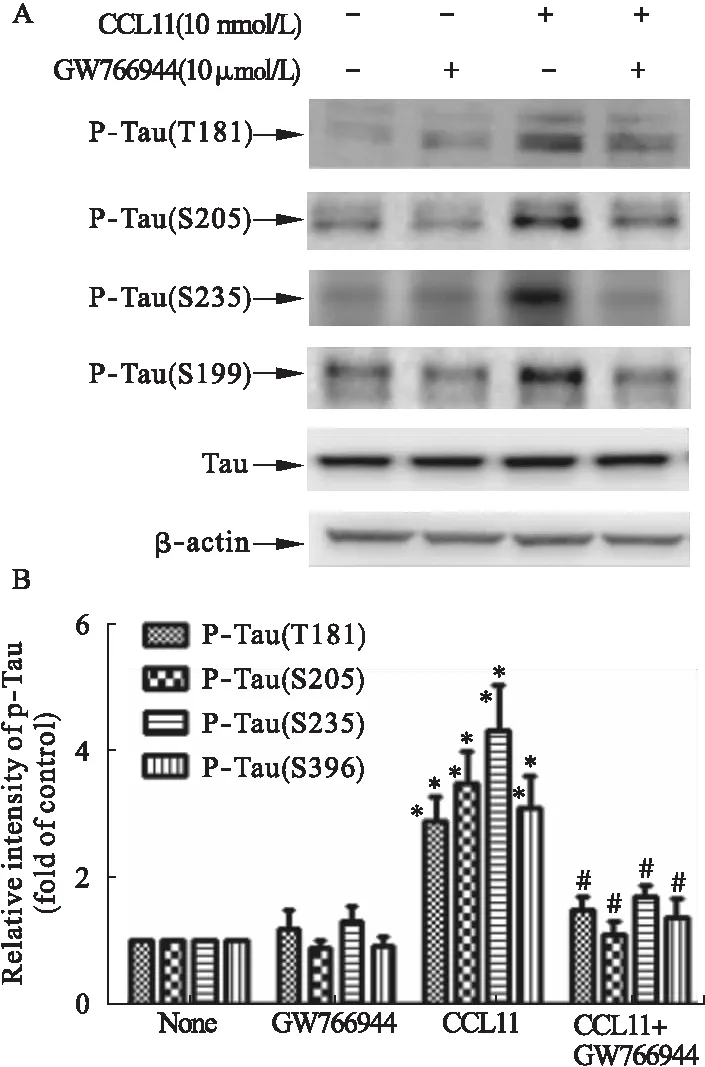
图5用CCL11(10nmol/L)处理海马神经元测试位点的tau磷酸化及逆转
注:A.海马神经元裂解物经过CCL11(10 nmol/L)处理10 min,加用和不加用GW766994(10 μmol/L)的培养物中,磷酸化tau在T181、T205、Ser235、Ser396位点的结果。B.不同磷酸化位点tau磷酸化的定量分析。量化数据表示为未处理样本值(时间零)的倍数。通过单向ANOVA分析差异。与同一治疗组的零时间点比较,*P<0.05,**P<0.01;与用载体处理的细胞相比,#P<0.05,##P<0.01

图6用CCL11(10nmol/L)处理海马神经元测试位点的tau磷酸化及逆转
注:A.在原代海马神经元培养物中CCL11对可溶性Aβ1-42的影响。Aβ水平通过ELISA方法,在经过载体或CCL11处理的细胞培养基中加入或者不加入GW766994的样本中测定。数据来自3个独立的实验,通过单向ANOVA分析差异。与未经任何处理的细胞相比,**P<0.01。与经过载体溶液和CCL11处理的细胞相比,##P<0.01。B.在原代海马神经元培养物中CCL11对可溶性Aβ1-40的影响。方法同上。尽管存在趋势,但CCL11处理对Aβ1-40没有观察到显著影响(P=0.077)

图7 CCL11处理导致神经元形态改变
在AD患者的大脑中,tau异常过度磷酸化,并且在这种改变的状态下,聚集成成对的螺旋状细丝,形成神经元纤维缠结。据推测,不同部位的tau蛋白过度磷酸化对其生物学功能及其致病作用有不同的影响。在Ser262、Thr231和Ser23位点的 tau过度磷酸化抑制其与微管的结合[17],在Ser199/Ser202/Thr205、Thr212、Thr231/Ser235、Ser262/Ser356和Ser422位点的超磷酸化,使tau转换成为一种抑制正常微管相关蛋白的抑制分子[18]。此外,新皮质中神经纤维缠结的密度与痴呆相关[19]。因此,开发合理的基于tau的治疗药物需要识别tau异常过度磷酸化的触发因素并理解其内在机制。趋化因子可能是涉及tau蛋白病理的重要因素。例如,CXCL1 通过CXCR2(CXCL1 受体)介导的ERK1/2激活触发原代神经元培养物中的tau 蛋白过度磷酸化。最近的一项研究进一步表明,用CXCL1处理长期神经细胞培养物导致tau以caspase-3依赖性方式切割。在本研究中,发现CCL11/CCR3在tau蛋白过度磷酸化中发挥作用。基于观察到纳摩尔浓度的CCL11直接诱导原代神经元培养物中的tau磷酸化,我们认为CCL11的年龄依赖性增加是AD的潜在危险因素。
除了tau蛋白病理外,AD的另一个重要病理标志是通过β-和γ-分泌酶裂解APP所产生的Aβ肽,聚集并在脑中形成的Aβ沉积物。γ-分泌酶将APP C-末端片段切割成2种主要形式的Aβ多肽Aβ40和Aβ42。Aβ42的相对量对于AD进展特别关键,其比Aβ40肽更容易聚集[20-22]。然而,在散发AD人群中,APP处理和Aβ产生的触发因素仍然很不明确。本研究结果表明,CCL11在原代神经元培养物中以CCR3依赖的方式诱导了显著的Aβ42增加和中等程度的Aβ40增加。
本研究结果表明,CCL11/CCR3在 Aβ产生,tau蛋白过度磷酸化和在突触丢失中起作用,同时CCL11诱导的蛋白激酶尤其CDK5和GSK3β的活化,也促进了上述有害作用的发生。GW766994作为一种特异性CCR3拮抗剂,显著逆转了上述过程,为AD治疗提供了可能的靶点。我们推测CCL11的这种增加是Aβ在大脑中产生的一个危险因素,并且早期干扰CCL11/CCR3通路可能延迟AD的发生。
在本研究中,我们证明CCL11培养导致了CDK5和GSK3β的显著活化。结果显示,CCL11增强了tau磷酸化和Aβ的产生,并与海马神经元培养物中的树突棘损失有关。CCR3的特异性拮抗剂GW766994逆转了上述全部过程。因此,拮抗CCR3介导的tau蛋白过度磷酸化,Aβ产生和突触损失可能会给AD带来治疗效果。
