HPLC法测定依达拉奉注射液中的有关物质
2019-04-01王连慧李俊广宋良伟张娜
王连慧,李俊广,宋良伟,张娜
(1.山东大学药学院,山东 济南 250100;2.山东罗欣药业集团股份有限公司,山东 临沂 276017)
依达拉奉注射液是由日本三菱制药公司开发并于2001年4月获日本厚生省批准上市,规格为20 mL:30 mg,商品名为Radicut[1],具有清除自由基和抑制脂质过氧化的作用,2003年进入中国,目前已有多家国产依达拉奉注射液上市。注射液有关物质检查是非常重要的一项质量控制指标,基于杂质谱进行本品有关物质研究目前尚无文献报道,本文结合本品所用原料药的杂质概况、制剂处方工艺、降解途径研究结果和相关文献资料对本品的杂质谱进行全面分析(见表1),在杂质分析的基础上,采用相应的杂质对照品对杂质分析方法进行系统的方法学验证,证明了所用的有关物质检测方法确实能有效地分离检出依达拉奉注射液中可能存在的杂质。
1 仪器与试药
1.1 仪器 XS105电子天平(梅特勒);岛津LC-20AT高效液相色谱仪(配置二极管阵列、紫外检测器)。
1.2 试药 依达拉奉注射液(山东罗欣药业集团股份有限公司,批号∶170301、170302、170303,规格20 mL:30 mg);依达拉奉注射液(日本田边三菱制药株式会社,A073);依达拉奉对照品(中国食品药品检定研究院);二聚体(深圳斯坦德);三聚体(深圳斯坦德);杂质P1(中国食品药品检定研究院)、杂质P3(深圳斯坦德);杂质1、杂质2均为自制;甲醇(色谱纯,禹王集团);磷酸二氢钾、磷酸、三乙胺(国药集团)均为分析纯试剂;水为纯化水。
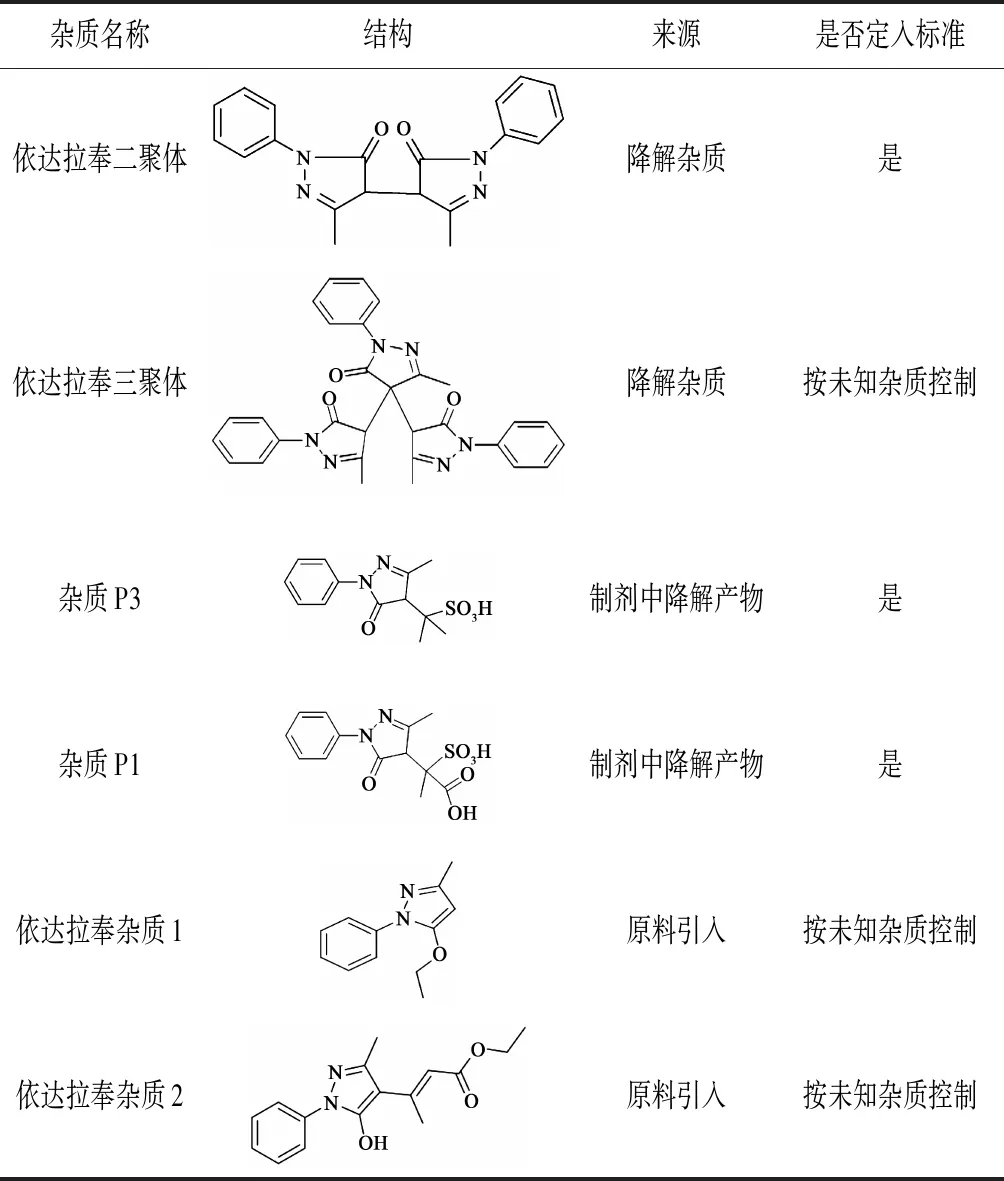
表1 依达拉奉杂质谱[2-3]
2 方法与结果
2.1 方法


表2 液相梯度洗脱表
2.1.2 溶液配制 取本品适量,加溶剂[0.05 mol·L-1磷酸二氢钾溶液(用磷酸调pH值至3.5)-甲醇(50∶50)]稀释制成每1 mL中约含依达拉奉0.5 mg的溶液,作为供试品溶液;精密量取供试品溶液适量,用溶剂稀释制成每1 mL中约含依达拉奉0.5 μg的溶液,摇匀,作为对照溶液。
2.2 结果
2.2.1 专属性
2.2.1.1 空白干扰试验 精密量取空白溶剂、空白辅料溶液和供试品溶液各20 μL,分别注入液相色谱仪,记录色谱图,空白溶剂及空白辅料对本品有关物质检查无干扰,各杂质峰与其他峰分离度均大于2.0,能完全分离。
2.2.1.2 强降解试验 取依达拉奉注射液在各苛刻试验条件(酸、碱、氧化、高温、光照)下进行破坏、降解,同时做空白辅料作为对照,按照本方法分别进行测定,结果见表3。
试验结果表明,本品在氧化条件下杂质个数及杂质含量增加明显,说明本品在氧化条件下有明显的降解,其他破坏条件下均较稳定;本方法辅料均无干扰,对各杂质分离良好,对测定无干扰,专属性符合要求,各个破坏条件下峰纯度符合要求,适于本品的有关物质检查。
2.2.2 检测限与定量限 精密称取依达拉奉与各杂质对照品适量,分别加甲醇溶解并稀释至信噪比约为10∶1,作为定量限溶液,连续进样6针,精密度良好;分别精密量取各定量限溶液3 mL,置10 mL量瓶中,加溶剂稀释至刻度,信噪比约为3∶1作为检测限溶液,分别精密量取各20 μL,注入液相色谱仪,记录色谱图,结果见表4。

表4 检测限与定量限试验结果
2.2.3 线性关系、校正因子的考察 精密称取依达拉奉和各杂质对照品适量,加甲醇溶解并用稀释剂制成一系列浓度的溶液,分别精密量取上述溶液各20 μL,注入液相色谱仪,测定峰面积。以浓度为横坐标,以峰面积为纵坐标进行线性回归,结果见表5。
结果表明:主成分与各杂质在一定的范围内,其浓度与峰面积成良好的线性关系,相关系数均大于0.999。杂质P1、杂质P3响应值与依达拉奉差异较大,对杂质P1和杂质P3的控制采用外标法。其他杂质校正因子均在0.9~1.1之间,采用不加校正因子的自身对照法测定其含量。

表5 线性试验结果
2.2.4 进样精密度试验 取依达拉奉及各杂质线性关系试验项下的中间浓度溶液,作为供试品溶液。精密量取20 μL,注入液相色谱仪,连续进样6次,记录色谱图,计算6次峰面积的RSD,结果见表6。

表6 精密度试验结果
结果表明主成分与各杂质RSD均小于2.0%,精密度符合要求,本法进样精密度良好。
2.2.5 重复性试验 取本品,按照“2.1”项下的方法测定,重复本试验6次,杂质P1、 P3按外标法以峰面积计算,其他杂质采用不加校正因子的自身对照法计算,试验结果见表7。

表7 重复性试验结果
由以上数据可得,重复本试验6次,所检出杂质个数一致及杂质含量基本一致,即本法重复性良好。
2.2.6 中间精密度试验 由不同人员在不同时间、不同仪器,分别进行重复性试验,杂质P1、 P3按外标法以峰面积计算,其他杂质采用不加校正因子的自身对照法计算,由不同人员在不同时间、不同仪器进行检验,杂质个数一致,杂质含量基本一致,本法中间精密度良好。
2.2.7 溶液稳定性 取依达拉奉注射液适量,用溶剂稀释制成每1 mL中约含0.5 mg的溶液,摇匀,作为供试品溶液;供试品溶液室温下(约25 ℃)放置,于0、2、4、6、8、10、12 h时,精密量取20 μL,分别注入液相色谱仪,记录色谱图。按面积归一化法计算,试验结果表明,本品室温放置12 h各杂质个数没有变化,杂质含量变化均小于0.02%,稳定性良好。
2.2.8 准确度 分别精密称取依达拉奉各杂质对照品各约5 mg,置同一100 mL量瓶内,加甲醇稀释至刻度,摇匀,作为混合杂质对照品贮备液,精密量取混合杂质对照品贮备液1.6 mL(80%)、2.0 mL(100%)、2.4 mL(120%)各3份,分别置9个20 mL量瓶内,精密量取注射液5 mL,分别置上述量瓶中,用溶剂稀释至刻度,摇匀,作为加样供试品溶液,精密量取混合杂质对照品贮备液2 mL置20 mL量瓶中,加溶剂稀释至刻度,摇匀,作为混合杂质对照品溶液;精密量取注射液5 mL,置20 mL量瓶中,用溶剂稀释至刻度,摇匀,作为供试品溶液,分别精密量取混合杂质对照品溶液、供试品溶液、加样供试品溶液各20 μL注入液相色谱仪,记录色谱图。按外标法以峰面积计算各杂质的回收率,结果见表8。

表8 准确度试验结果
试验结果表明:各杂质平均回收率均在90%以上,RSD均小于10%,表明本法的准确度可靠。
2.2.9 耐用性试验 为考察本方法对条件发生微小变化的耐受程度,采用不同厂家仪器、不同品牌色谱柱、不同柱温(30±5)℃、不同流速(1.0±0.1)mL·min-1、不同波长(243±2)nm、不同pH(7.0±0.2)等因素对方法的耐用性进行考察,分别精密量取供试品溶液、混合溶液20 μL,注入液相色谱仪,记录色谱图。试验结果表明各杂质峰与其他成分峰均达到基线分离;杂质检出个数和总量基本一致,本方法耐用性良好。
2.2.10 样品测定及结果 分别取本品3批及原研制剂1批,加溶剂稀释制成每1 mL中约含0.5 mg的溶液,作为供试品溶液;精密量取供试品溶液适量,用溶剂稀释制成每1 mL中约含0.5 μg的溶液,摇匀,作为对照溶液。另取杂质P1和杂质P3对照品各适量,分别加少量甲醇溶解后,用溶剂稀释制成每1 mL中含P1约2 μg、P3约1 μg的溶液,作为对照品溶液。照上述确定的色谱条件进行测定,结果见表9。

表9 样品测定结果
3 讨论
3.1 检测方法的选择 已有文献报道[4-8]采用HPLC法测定依达拉奉有关物质,但没有针对杂质谱进行依达拉奉注射液有关物质的研究,本文针结合本品所用原料药的杂质概况、制剂处方工艺、降解途径研究结果和相关文献资料对本品的杂质谱进行全面分析,在杂质谱分析的基础上,采用相应的杂质对照品对杂质分析方法进行系统的方法学验证,证明了所用的有关物质检测方法确实能有效地分离检出依达拉奉注射液中可能存在的杂质。
3.2 与原研制剂杂质谱的比较 采用经过验证的有关物质检查方法对自制制剂和原研制剂进行杂质谱比较,由表9试验结果表明自制制剂的杂质个数少于原研制剂,杂质含量与原研制剂基本一致,说明自制制剂有关物质控制水平与原研制剂相当。
