Pt/Au(111)表面合金电化学稳定性的第一性原理研究
2019-03-29宋明可刘迅林谢先茂邓磊汤剑锋
宋明可,刘迅林,谢先茂,邓磊,汤剑锋*
(湖南农业大学 理学院,湖南 长沙 410128)
工业革命以来,化石能源储量不断减少,大气中温室气体不断增加,新能源的开发与利用引起了许多人的关注。聚合物燃料电池(PEFCs)拥有清洁和可持续发展的优点,是解决能源危机的方法之一[1]。阴极催化剂是决定PEFCs性能、寿命和成本的关键材料。现在市面上的常用催化剂还是以Pt材料为主[2~4],在苛刻电化学环境中,操作不当等因素使得Pt催化剂逐渐溶解,其耐久性有待提高。
人们通常通过改变衬底、形貌或者掺杂等方式来增强Pt催化剂的稳定性。Seung Hyo Noh等[5-6]设计了 Pt/Co、Pt/Ni、Pt/Cu、Pt/Cu/Ni等一系列的Pt肤合金纳米粒子,使得表面的Pt肤更难以溶解,相比于纯Pt催化剂具有更好的稳定性。黄小青等[7]将不同的过渡态金属(V、Cr、Mn、Fe、Co、Mo、W、Re) 掺杂在Pt3Ni纳米粒子表面,发现Mo-Pt3Ni/C的ORR性能最好,比活性为10.3 mA/cm2,质量活性为6.98A/mg,比商用Pt提高81倍和73倍,同时也增强了该合金的稳定性。这些方法需要将其他金属掺杂到Pt催化剂的次表层或里层,有的甚至要掺杂几种金属,这在实际中操作比较复杂,需要寻找一种更为简单的掺杂金属。
R.Jinnouchi等[8-9]采用密度泛函理论(DFT)计算发现,Pt纳米催化剂中存在从颗粒边缘开始溶解的逐层溶解机理。随后,他用Au原子替换纳米粒子边缘的Pt原子后发现,虽然Au原子周边的Pt原子溶解电位减小,但是该催化剂的最低溶解电位得到提高,即整体的稳定性增强,并且对(111)面铂吸附位点的催化活性没有影响。这说明用少量的Au掺杂来增强Pt催化剂的稳定性是可行的。本研究旨在通过研究Pt/Au(111)表面合金这一模型催化剂的表面结构及其溶解电位,以定量揭示表面Au含量与排布方式对表面Pt原子电化学稳定性的影响。
1 模型与方法
本次计算使用的DFT软件是VASP,价电子与离子核之间相互作用使用PAW势描述,交换相关函数采用GGA-PBE。在每个K点上使用波函数基组且动态截断能为450eV。布里渊区在K点上的总和使用Monkhorst-Pack方法,设置为5×5×1。表面结构优化的能量收敛标准为1×10-4eV/atom,张力收敛标准为0.02eV/Å。
所计算的表面为Pt(111)salb模型,采用优化后Pt块体的晶格常数(3.977Å),共构建4层,每层16个原子,最上层加入12Å的真空层以避免周期性边界条件引起的表面间相互作用。结构优化过程中固定下面2层原子,允许上面2层原子弛豫。Pt/Au(111)表面合金是将Pt(111)salb模型中的Pt原子替换成金Au原子来构建的。模型如图1所示。图1(a)表示Pt(111)slab模型,图1(b)表示Pt/Au(111)模型,其中蓝色原子表示Pt原子,金色和灰色原子分别代表中心Pt原子的表面第一近邻和第二近邻的替位Au原子。

图1 (a)Pt(111)slab模型; (b)Pt/Au(111)slab模型
2 结果与讨论
2.1 Pt/Au(111)表面合金的热力学稳定结构
应用DFT方法计算出体系的形成能,能够预测一定的合金组分或分布方式下该合金的热力学稳定结构。分别计算了不同Au含量和相同Au含量但不同分布方式两种情况下的表面合金形成能。由n个Pt原子和m个Au原子组成的PtnAum(111)表面单原子形成能Ef(PtnAum)的计算公式如下:

在式 (1)中,E(PtnAum)、E(Ptn+m)、E(Aun+m)分别代表用DFT计算PtnAum(111)合金、Ptn+m(111)合金和Aun+m(111)合金的总能,x代表该合金的表面原子数。

图2 Pt/Au(111)表面合金的组态与形成能的关系
图2描述了Pt/Au(111)表面合金的组态与形成能的关系。其中黑色方框表示Au取代在表层位置的Pt/Au(111)合金,实心黑色方框表示形成能最低的结构,橙色方框表示Au取代在次表层位置的Pt/Au(111)合金,实心橙色方框表示形成能最高的结构。根据热力学理论,在确定Au成分的情况下,位于最下方的黑色实心方框是热力学最稳定的组态。通过对Pt/Au(111)合金仔细分析,Pt则更倾向于里层。例如在Au占Pt/Au(111)合金表面的含量为25%时,Au分布在次表面的表面单原子形成能远高于Au分布在表层的状态。在Au占Pt/Au(111)合金表面的含量为6.3%、12.5%、18.8%也是相同的情况,并且随着次表层Au含量的增多形成能越来越高。这是由于Au的表面能远小于Pt,从而更倾向于占据合金的表层位置。同时还观察到:对于确定Au含量的Pt/Au(111)合金(只考虑Au替换在表层的情况),形成能最低的结构(黑色实心方框)对应Au在表面偏析且团聚,相反的是,形成能最高的结构(最上的黑色空心方框)为Au在表面分散,形成稀固溶体。例如在表面Au含量为18.8%、25%的Pt/Au(111)合金处,偏析且团聚状态的形成能分别为-0.201eV/atom、-0.276eV/atom,稀固溶体的形成能分别为-0.093eV/atom、 -0.084eV/atom, 相 差 有 -0.1~-0.2eV/atom。不同表面Au含量的情况下具有相同的特征,说明Au倾向于在Pt/Au(111)表面偏析且高度聚集,这源于Au与Pt之间的形成能为正,与块体相图一致。另外,还能看到,在合金表面Au含量为25%时为-0.276eV/atom、50%时为-0.661eV/atom、75%时为-1.127eV/atom、100%时为-1.439eV/atom,随着表面Au含量的增加,形成能越来越低。这说明在Pt中掺杂Au原子,Au会自发占据Pt金属的表面并成团,随着随着表面Au含量的增加将覆盖整个表面。
2.2 Pt/Au(111)表面合金的电化学稳定性
虽然Au原子很稳定,但是它的掺杂会对周边的Pt原子的稳定性造成影响。这也意味着当Pt(111)表面掺杂了Au后,其表面的溶解电位可能变得分布不均匀。溶解反应将从溶解电位最低的位置开始,因而获取表面参杂后铂的溶解电位分布十分重要。分别计算了该合金表面Pt原子在其周围第一近邻(图1(b)中黄色原子位置)与第二近邻(图1(b)中灰色原子位置)不同Au原子含量下的溶解电位。溶解电位UPt计算公式[8-9]如下:
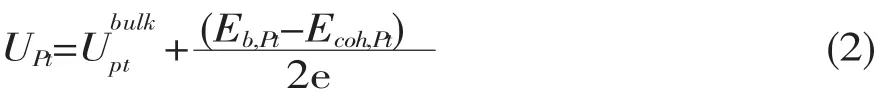

在式 (3) 中 E(Ptn-1Aum)、E(Pt)、E(Ptn-1Aum)分别代表用DFT计算Ptn-1Aum(111)合金、单个Pt原子和PtnAum合金的能量。
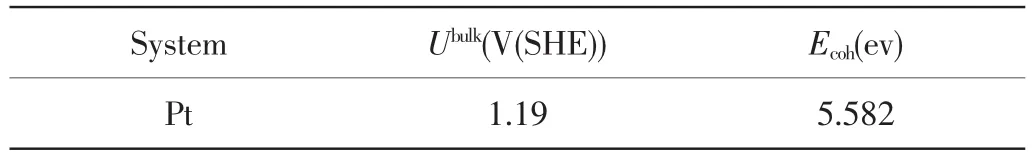
表1 块体铂的内聚能与溶解电位
图3描述了Pt/Au(111)合金表面中Pt原子周围的第一近邻Au原子对其溶解电位的影响。图4描述Pt/Au(111)合金表面中Pt原子第二近邻的Au原子数对其溶解电位的影响。能够观察到,随着Pt原子周围第一近邻Au原子的增加,溶解电位越来越小,这说明合金化效应对于Pt原子的电化学稳定性影响很大。此外,随着第二近邻Au原子的增加,中心Pt原子的溶解电位几乎不变,差值在0.001V以内。这说明第二近邻的Au原子对Pt的稳定性影响很小,只需要考虑第一近邻位置的影响。

图3 Pt/Au(111)合金表面中Pt原子周围的第一近邻Au原子和其溶解电位的关系

图4 Pt/Au(111)合金表面中Pt原子周围的第二近邻Au原子和其溶解电位的关系
在图3中,进一步对NAu与UPt进行了线性拟合,相关系数r的绝对值为0.98,即二者具有非常好的线性关系。这意味着可以通过该关系来较精确的评估铂纳米结构的表面组态与电化学稳定性关系,为设计更稳定的Pt/Au催化剂提供了理论指导。
3 结论
本文运用密度泛函理论对Pt/Au(111)合金催化剂的稳定性进行了研究,发现Au掺杂在表层的形成能比在次表层和块体内部的形成能更低,即Au更容易偏析到表面并聚集成团,说明了用Au来修饰Pt催化剂表面的热力学可行性。在Pt/Au(111)合金中Au对第二近邻及更远近邻的Pt的溶解电位影响可以忽略不计,而第一近邻的Au原子数对其溶解电位的影响有非常好的线性关系,且可通过此关系来精确预测具有一定表面合金组态的Pt/Au催化剂的电化学稳定性。
