Hepatic encephalopathy: Lessons from preclinical studies
2019-03-21LuizaCiogliaDiasLimaAlineSilvaMirandaRodrigoNovaesFerreiraMileneAlvarengaRachidAnaCristinaSimesSilva
Luiza Cioglia Dias Lima, Aline Silva Miranda, Rodrigo Novaes Ferreira, Milene Alvarenga Rachid,Ana Cristina Simões e Silva
Abstract Hepatic encephalopathy (HE) is a major complication that is closely related to the progression of end-stage liver disease. Metabolic changes in advanced liver failure can promote cognition impairment, attention deficits and motor dysfunction that may result in coma and death. HE can be subdivided according to the type of hepatic injury, namely, type A, which results from acute liver failure, type B, which is associated with a portosystemic shunting without intrinsic liver disease, and type C, which is due to chronic liver disease. Several studies have investigated the pathogenesis of the disease, and most of the mechanisms have been explored using animal models. This article aimed to review the use of preclinical models to investigate HE. The most used animal species are rats and mice. Experimental models of type A HE include surgical procedures and the administration of hepatotoxic medications, whereas models of types B and C HE are generally surgically induced lesions in liver tissue, which evolve to hepatic cirrhosis. Preclinical models have allowed the comprehension of the pathways related to HE.
Key words: Hepatic encephalopathy; Acute liver failure; Preclinical studies; Hepatic cirrhosis; Neuroinflammation; Hyperammonemia
INTRODUCTION
Hepatic encephalopathy (HE) is defined as a neuropsychiatric syndrome that occurs in patients with acute or chronic liver diseases[1]. Clinically, it is characterized by a spectrum of symptoms, including cognition impairment, altered levels of consciousness that may progress to coma and death[2]. The risk of mortality has usually been related to higher grades of HE, and the incidence of this disease is increased in patients with frequent infections[3,4]. Patients with HE are greatly impacted in terms of quality of life and the high costs caused by increased healthcare utilization[5,6].
The pathophysiology of HE is not completely understood, and several studies have shown biochemical disturbances, with elevation of serum ammonia levels and increased oxidative stress in blood, alterations of neurotransmission systems,development of brain edema, astrocyte swelling and inflammation[8]. A wide range of animals, including dogs, goats, pigs, rabbits, guinea pigs, rats and mice, have been used for better understanding the mechanisms underlying HE pathogenesis and for the development of novel therapeutic agents for HE, providing a basis for future clinical research. Therefore, the objective of this brief review is to discuss relevant aspects of the HE pathogenesis and the current animal models employed, which may closely resemble the disease in humans.
HEPATIC ENCEPHALOPATHY
HE is a central nervous system (CNS) dysfunction resulting from acute or chronic liver failure that leads to a wide range of neuropsychiatric manifestations[7]. Clinically,patients with HE may present sleep-wake cycle disturbance, personality changes and cognitive, motor activity and coordination dysfunctions, which ultimately progress to stupor, coma and death. Importantly, HE often affects health-related quality of life,clinical management strategies, liver transplant priority, and patient survival[8].
According to the American Association for the Study of Liver Disease (AASLD)updated guidelines, HE should be classified as type A, B, or C based on the underlying disease. Type A (acute) results from an acute liver failure (ALF), while type B (bypass) is associated with a portosystemic shunting without intrinsic liver disease and type C (chronic) is the consequence of cirrhosis[7]. The incidence of ALF is low, affecting approximately 2000 people per year in the United States or Europe[8,9].However, the mortality of ALF is high and is mostly attributed to fast progression to HE, leading to cerebral edema, increased intracranial pressure and cerebral herniation[8,9].
In contrast, chronic liver diseases are highly prevalent, affecting approximately 5.5 million individuals only in the United States. Approximately 80% of patients with cirrhosis will develop a less severe form of HE, known as minimal HE (MHE), which is characterized by mild cognitive impairment, attention deficits, psychomotor slowing and impaired visuomotor and bimanual coordination[10]. MHE is detected only by employing psychometric or neurophysiological tests and is considered an important predictive factor for the development of HE since 30%-40% of cirrhotic patients progress to this later condition[10,11]. Apart from its educational and social impact, HE also carries a significant economic burden. For instance, in 2009, in the United States, HE led to approximately 22931 hospitalizations, with an average cost of each stay ranging from 46663 to 63108 USD[8].
The broad spectrum of HE, especially regarding the type of underlying disease,severity of clinical manifestations, and precipitating factors (e.g., infections,gastrointestinal bleeding and drug toxicities) should be considered to understand the pathophysiological mechanisms of HE as well as to identify potential therapeutic targets[12].
HEPATIC ENCEPHALOPATHY PATHOPHYSIOLOGY
HE is a complex condition who’s cellular and molecular mechanisms remain to be fully elucidated. Over the past decades, some hypotheses have been proposed,including neurotransmitter system dysfunction, impaired energy and lactate metabolism and oxidative stress (for review see[1,13]). However, the hyperammonemia and the neuroinflammation hypotheses have been the mostly recognized ones and will be briefly revisited in the current review. Figure 1 shows the main pathophysiological features of the available models of HE.
Hyperammonemia
Currently, increased systemic and brain levels of ammonia are the main factors implicated in HE pathogenesis. Under physiological conditions, the ammonia,resulting from nitrogenous compounds, such as proteins metabolized by gut microflora, is metabolized in the liver via the urea cycle, forming urea, which is mainly excreted by the kidneys. ALF, portosystemic shunting or chronic liver disease can impair liver function, leading to increased levels of ammonia in the blood[14].
Ammonia metabolism in the liver depends on phosphate-activated glutaminase(PAG), which catalyzes the hydrolysis of glutamine to produce glutamate, energy,nucleotide synthesis and ammonia. PAG has two isoforms, the hepatic type (L-PAG),restricted to the liver, and the kidney-type (K-PAG), found in the kidney, brain and enterocyte villi, especially in the small intestine. Interestingly, PAG activity in the intestine has been associated with increased systemic levels of ammonia during liver cirrhosis and seems to play a major role in the pathogenesis of HE[15,16].
As the levels of ammonia increase systemically, the molecule crosses the bloodbrain barrier and starts to be metabolized in the CNS[14]. The ammonia detoxification in the brain requires its incorporation into glutamine by the action of the enzyme glutamine synthetase, which is present only in astrocytes. The glutamine accumulation in astrocytes as a result of ammonia detoxification results in increased water entry and osmotic forces, ultimately inducing astrocytes to swell and causing cytotoxic edema[17]. The impact of hyperammonemia on astrocyte function in response to HE remains to be fully elucidated. However, it has been reported that, apart from increasing oxidative stress and osmotic pressure, hyperammonemia may also influence inflammatory and signal transduction pathways[18,19], gene expression and neurotransmitter release[20]as well as posttranslational protein modifications[21].
Although a great deal of attention continues to be focused on ammonia as the main toxin related to HE pathogenesis, there is evidence that patients with elevated levels of systemic and local ammonia may not present HE symptoms; additionally, the ammonia concentration is not always consistent with the severity of HE in cirrhotic patients[22,23]. Moreover, ammonia-lowering agents, includingL-ornithine,L-aspartate and lactulose, have so far been of limited value in preventing HE in ALF and in cirrhosis[24-26], supporting a role for other factors alone or in association with ammonia in the development of HE. For instance, the effect of the glycerol phenylbutyrate(GPB), approved by the Food and Drug Administration in 2013 for the treatment of urea cycle disorders, was investigated in a randomized, double-blind, placebocontrolled phase II trial with cirrhotic patients who experienced two or more HE events in the last 6 mo. The GPB acts as an ammonia-lowering agent by producing phenylacetyl glutamine, which is excreted in urine. GPB treatment in cirrhotic patients decreased plasma levels of ammonia, the proportion of patients who experienced HE and hospitalizations due to HE. These findings supported the involvement of ammonia in HE pathophysiology and the potential therapeutic role of GPB[27].
Neuroinflammation
In addition to the ammonia hypothesis, brain inflammation, also known as neuroinflammation, is thought to be a major component in the development of HE.Clinical and experimental evidence of activation of microglia, the brain resident immune cells, in response to ALF and cirrhosis extensively supported the neuroinflammation hypothesis[28-32]. For instance, increased expression of the major histocompatibility complex class II antigen marker CD11b/c (also termed OX-42), an indicator of microglial activation, was found in an ALF model following liver ischemia in rats[30]. Importantly, the administration of minocycline, a potent inhibitor of microglial activation, attenuates the encephalopathy grade and prevents brain edema in the same ALF experimental model[32]. Corroborating these findings,increased expression levels of microglial activation markers, including human leukocyte antigen DR (CR3/43) and ionized calcium-binding adaptor molecule 1 (Iba-1), were found in the CNSs of patients with ALF associated with viral hepatitis[29]and in liver cirrhosis[31], respectively.
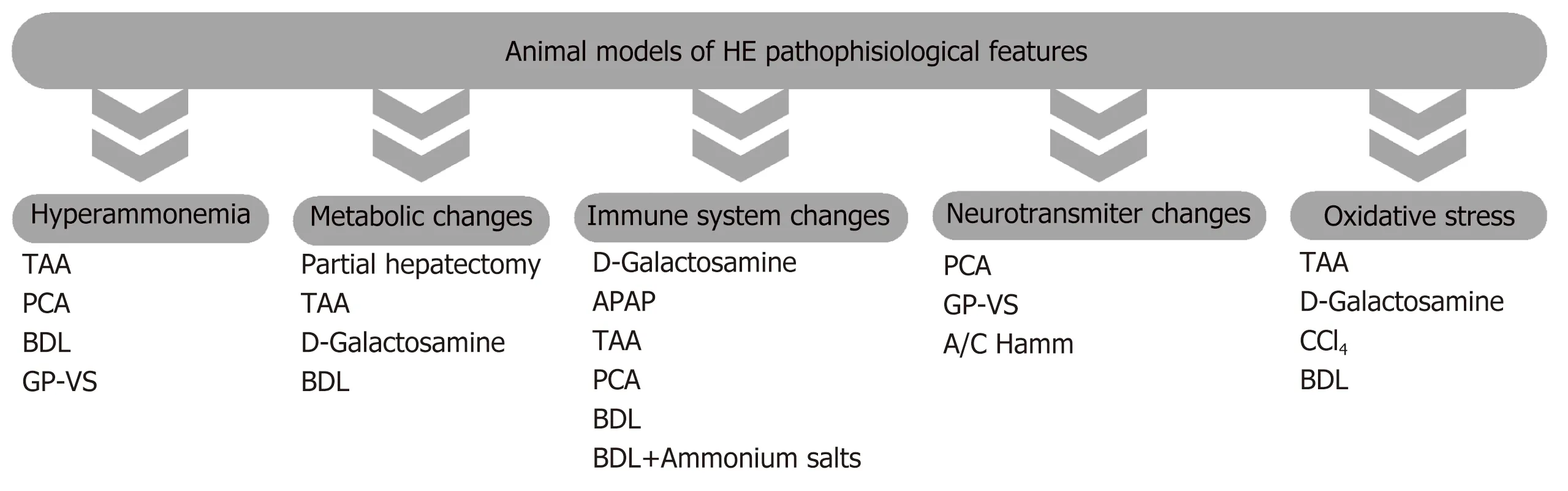
Figure 1 Main pathophysiological feature of the available models of hepatic encephalopathy. APAP: Acetaminophen; A/C Hamm: Acute/chronic hyperammonemia; BDL: Bile duct-ligated; CCl4: Carbon tetrachloride; GP-VS: Graded portal-vein stenosis; PCA: Portosystemic anastomosis; TAA: Thioacetamide.
Microglial activation has often been associated with increased release of cytokines and chemokines, which are also implicated in the pathogenesis of neurodegenerative and neuropsychiatric diseases[33,34]. The first evidence of upregulation of inflammatory molecules in HE was obtained in a study of patients with ALF due to acetaminophen overdose. The levels of inflammatory cytokines (TNF, IL-1β and IL-6) were measured in blood samples from an artery and a reverse jugular catheter. Increased arterial levels of cytokines correlated with intracranial hypertension. Brain cytokine efflux was noted, indicating brain cytokine production in these patients[35]. Supporting data were also provided by several studies employing pharmacological and nonpharmacological liver failure models in rodents, which also showed increased levels of inflammatory cytokines (TNF, IL-1β and IL-6) and chemokines, such as CXCL-I, CCL2, CCL3, CCL5 and CX3CL1[30,32,36-38]. Importantly, anti-inflammatorybased strategies attenuated cognitive decline and motor activity impairment,supporting the involvement of neuroinflammation in HE pathophysiology[39-42]. It is worth mentioning that ammonia alone is capable of inducing microglial and astrocyte activation, leading to increased expression of inflammatory cytokines, such as IL-1β and IL-6. This finding suggests that hyperammonemia may trigger neuroinflammation in HE in a synergistic manner. However, a direct link between both mechanisms is still missing[43].
PRECLINICAL MODELS
Choice of animal species
In 2008, a commission formed by members of ISHEN (International Society of Hepatic Encephalopathy and Nitrogen Metabolism) gathered in Padua, Italy to establish guidelines for HE animal models. Due to the variety of etiologies of hepatic dysfunction and the many factors that may influence the development of neurological symptoms, HE is considered difficult to reproduce faithfully in animals. Table 1 summarizes the main advantages and disadvantages of the available HE models.Currently, there is no ideal animal model to resemble hepatic failure caused by ethanol, virus or acetaminophen, the most common etiologies in human beings[44].
The availability of animal models is indispensable for studying the mechanisms of diseases and possible therapies[45]. Many animal species have been used for the investigation of HE, including dogs, goats, pigs, rabbits, guinea pigs, rats and mice.Large animals are advantageous for neurological examinations and the availability of biological samples, such as blood samples, body fluids and biopsies but are rarely used due to the cost of the animals, the maintenance involved and ethical concerns.The most common species currently used in HE models are rats and mice, mainly due to the accessibility of molecular and anatomical studies and the availability of literature on behavioral, pathological and biochemical methods and findings. Other advantages of these species are the complete characterization of the genome, easy availability of antibodies and molecular probes and low costs for obtaining and maintaining the animals[44].
Animal HE models of ALF (type A)
According to the HE guidelines established by ISHEN, an animal model of HE in ALF has to essentially reproduce the clinical picture to facilitate staging of encephalopathy and to show the progression of symptoms, including brain edema and its complications (i.e., intracranial hypertension and brain herniation). The model should also be capable of being reversed, with high concentrations of ammonia and glutamine in the brain, classical hepatic and brain pathology and minimal hazards to personnel from toxins and infectious agents. All animal models of ALF produce hypothermia and hypoglycemia, making it essential to control the temperature and glucose levels and to provide supportive care to the animals. A brief description of ALF models is presented in Table 2.
Pharmacological models
Several hepatotoxins have been extensively used in the development of animal models of ALF[46]. The main hepatotoxic substances used to cause ALF in animals include galactosamine, acetaminophen, azoxymethane (AOM) and thioacetamide(TAA)[44]. Although the administration of carbon tetrachloride (CCL4) has been primarily described as a type A HE model, the ALF and behavioral changes in this model are not commonly observed. However, the ability of this model to induce liver cirrhosis makes it a valuable tool for studying type C HE[44]. Thus, in the current review, the CCL4model will be discussed as a Type C HE model.
Thioacetamide:Until 1943, TAA was used as fungicide in orange crops. In 1948, it was discovered that chronic administration of the substance led to liver cirrhosis and hepatocellular carcinomas. TAA causes hepatocellular necrosis after biotransformation to an active metabolite via the flavin adenine dinucleotide monooxygenase pathway, resulting in the formation of TAA-S-oxide[47]. TAA reduces antioxidant activity and enhances lipid peroxidation in the liver, leading to oxidative stress and cellular necrosis[48]. The most common forms of administration of TAA are oral (in drinking water) and intraperitoneal injection[49]. TAA has been extensively used to induce ALF in rats and mice, producing encephalopathy, metabolic acidosis,high transaminases, abnormal coagulation and histological centrilobular necrosis. The TAA model of HE shows good reproducibility and well-described hepatic and cerebral changes[50-52]. Chronic administration can produce hepatic cirrhosis[49,53,54].
D-Galactosamine:D-Galactosamine (D-Gal) is an amino sugar that is metabolized in the liver, causing consumption of uridine nucleotides and blockade of transcriptional factors in the liver. The hepatic failure resulting from D-Gal administration has been,at least in part, associated with the production of uridine-containing compounds during hepatic biotransformation[55]. The administration of D-Gal in rats was described by Keppler et al[56]as an acute hepatic failure model that induced encephalopathy and increased aspartate transaminase, prothrombin time, ammonia and intracranial pressure. The same model was described by Blitzer et al[57]in rabbits and by Sielaff et al[58]and Diaz Buxo et al[59]in dogs.
There are differences in species susceptibility. Mice are resistant until high doses are administered, while rats are sensitive to D-Gal-induced hepatic failure[60]. D-Gal and acetaminophen models cause liver failure, although the development of HE is variable. Both models are difficult to reproduce and have poorly characterized cerebral pathology[44].
Acetaminophen:Acetaminophen is known as APAP (in the United States) and Paracetamol (in Europe and other areas of the world). Acetaminophen has been widely used as an antipyretic and/or analgesic since 1955, particularly because it is easily accessible in various formulations as an over-the-counter medication. However,acetaminophen is one of the most common causes of ALF, accounting for more than 60% of all cases in the United States[61].
The metabolism of APAP occurs in the liver. Under normal circumstances, the medication undergoes biotransformation by a combination of glucuronidation and sulfation and is then excreted by the kidneys. When excessive amounts of acetaminophen are administered, these metabolic pathways are saturated, and APAP is metabolized by the P450 cytochrome oxidase enzyme system, leading to the formation of a toxic electrophile, N-acetyl-p-benzoquinoneimine (NAPQI). NAPQI produces cell injury unless conjugated with endogenous glutathione. NAPQI is supposed to disrupt mitochondrial calcium flux, resulting in cell damage by the production of free radical oxygen species, hydroxyl radicals, nitrites and nitrates. The cascade is amplified by the activation of Kupffer cells and the release of cytotoxic mediators (e.g., cytokines and free radicals), leading to apoptosis and cell necrosis[62-65].
Miller et al[66]reported a model of APAP-induced liver failure in pigs. Animals exhibited metabolic acidosis, encephalopathy, coma, increased transaminase levels and histological evidence of severe centrilobular coagulative necrosis. However,changes produced by APAP administration were variable according to animal species used, as was described by Gazzard et al[67], who showed a variable clinical outcome in greyhound dogs.
Later, Francavilla et al[68]induced ALF by APAP administration for the first time in beagles. The authors compared the routes of administration: intravenous,intramuscular and subcutaneous. APAP toxicity was very variable in different animal models, and the incidence of sudden death due to methemoglobinemia wasfrequently observed. This drug exhibited cardiotoxicity and nephrotoxicity, which was associated with acute lung injury[69-71].
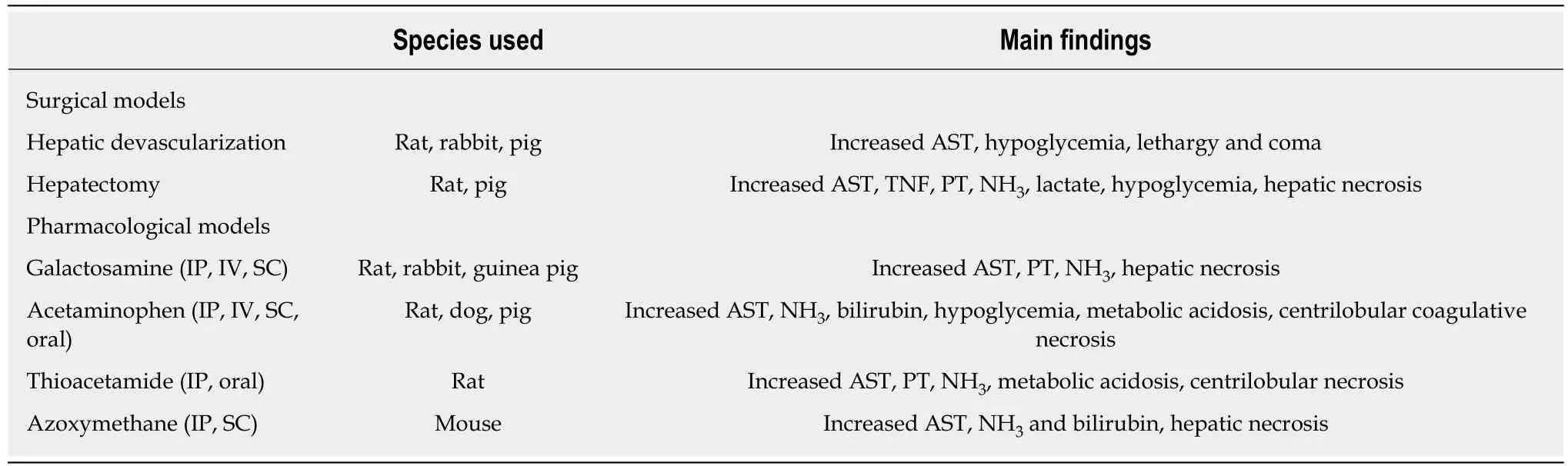
Table 2 Brief description of type A animal models of hepatic encephalopathy
Azoxymethane:AOM is an active metabolite of the cycad palm nut found on the island of Guam. AOM is a potent hepatotoxin that induces ALF in mice in a dosedependent manner. Liver toxicity has also been reported in humans, livestock and rats following the ingestion of Guam cycad palm nuts due to AOM toxicity. The AOM model was first described as a model of hepatotoxin-induced liver failure and HE by Matkowskyj et al[72]. This model leads to encephalopathy, cerebral edema, elevated brain ammonia and unbalanced amino acid levels. This model also shows characteristic pathologic aspects[28].
The AOM model generates microvesicular steatosis, dilation of hepatic sinusoids,and hepatocyte necrosis in addition to elevations in serum transaminases and bile acids, with the largest increases observed when mice progressed toward coma[73].
Surgical models
The surgical models can be divided into variations of partial and total hepatectomy and partial and complete devascularization of the liver[46].
Hepatic devascularization:Rappaport was the first to describe a devascularization model, in 1953[74]. The model is usually produced by a portocaval anastomosis (PCA)with subsequent hepatic artery ligation (HAL). ALF can be reversible by the occlusion of the hepatic artery for only a short period of time. There is no blood lost. The presence of necrotic hepatic tissue is comparable to ALF in humans[45]. Hypoxic insult results in dysfunction of the mitochondrial respiratory chain, which, in turn, reduces ATP levels due to impaired oxidative phosphorylation, interferes with the intracellular calcium homeostasis and activates enzymes responsible for protein, lipid and DNA damage. After hypoxic injury, the reperfusion process induces more damage in liver tissue[75-77].
Hepatectomy:Hepatectomy is considered a model of postoperative liver failure(POLF) that can accurately reproduce all neurological and metabolic changes as a consequence of extensive liver resections in humans[78]. Almost 100% survival and intense regeneration occur in partial hepatectomy that removes approximately 70% of the liver in rats and pigs[79-81]. In contrast, progressive necrosis follows partial resections of the liver, as reported by Panis et al[82]. Among other disadvantages of partial hepatectomy are the lack of consistency of injury degree and the increased intraoperative blood loss. Additionally, the surgery often hampers the animal’s capability to immediately restore drink and food consumption, which may lead to severe hypoglycemia and death[83].
The altered physiological state is attributed to increased total blood flow through the remnant liver tissue, leading to flow injury and damage to sinusoidal endothelial cells, activation of Kupffer cells and release of inflammatory cytokines[84]. This model is also associated with increased levels of aspartate transaminase (AST), tumor necrosis factor (TNF) and hypoglycemia[85].
The “anhepatic” model is made by the total removal of the liver. In clinical practice,total hepatectomy has been performed only in cases of very severe ALF, pending the arrival of a donor liver, which follows the removal of a “toxic” organ[86,87]. Overall, the anhepatic model seems not to be suitable for evaluating therapies as an artificial liver support for ALF. Moreover, due to the lack of the liver, this model has very poor reversibility[46].
Animal models of HE types B and C
An ideal model of HE classified as type B or C should have some essential features,including a precipitant factor and neuropathological findings of HE, such as symptoms of encephalopathy, ranging from MHE to coma. Additionally, increased brain ammonia/glutamine along with low-grade brain edema and Alzheimer Type II astrocytosis at advanced stages of HE might also be observed. These models should exhibit clinical responses to established treatments[29]. Table 3 displays the animal models of HE associated with portosystemic shunting (type B) and chronic liver disease (type C).
Type B hepatic encephalopathy
As previously mentioned, animal models of type B HE are based on portal-systemic shunting and include the following approaches.
Portacaval anastomosis:PCA is the most common animal model used in the study of chronic HE. The basis of this model is the creation of a portal-systemic shunt (end-toside PCA) that mimics the situation induced in cirrhosis by collateral circulation[45].
It is known that the deviation of portal blood decreases the total hepatic blood flow,depriving the liver of oxygen and hepatotrophic factors from the portal vein that are necessary for metabolic processes. These phenomena induce hepatic parenchyma atrophy. Among other factors, the high levels of ammonia, as a consequence of reduced hepatic metabolism, contribute to HE. The severity of neurological manifestations depends on the intensity of liver injury. Animals submitted to PCA have increased brain ammonia/glutamine, altered circadian cycle, hypokinesia and reduced memory and learning ability[88].
Congenital portacaval shunts:Dogs and cats with congenital portacaval shunts are considered naturally occurring models of type B HE. These animals develop psychomotor dysfunction, abnormal motor signs, altered day-night rhythms,hyperammonemia, and hepatic dysfunction[89].
Graded portal vein stenosis:Graded portal vein stenosis provides a MHE model in rats. This procedure is easier to perform than the end-to-side portacaval anastomosis.Alterations associated with this model are hypoactivity, altered circadian cycle and increased brain ammonia/glutamine[44].
Type C hepatic encephalopathy
Animal models of type C HE should lead to decompensated liver cirrhosis. It is worth noting that there is currently no ideal animal model of type C HE[44]. Some models that resemble human hepatic cirrhosis have been employed, as described below.
CCl4:CCl4was extensively used in the 1970s as a model of acute hepatic failure, but the species variation was significant, and it was difficult to reproduce. It has been used more recently in the study of liver cirrhosis[90,91]. CCl4is metabolized by cytochrome P-450, generating free radicals, causing lipoperoxidation, increased hepatic membrane permeability, tissue fibrosis and hepatic failure. This model does not seem to produce many behavioral changes, except in cases of advanced cirrhosis[92]. It is used in the study of astrocytic response at the level of RNA synthesis.Inconsistent lesions animal to animal may represent a major disadvantage. Moreover,the presence of ascites may limit neurobehavioral assessments[44].
Bile duct ligation:The ligation of the common bile duct ligation (BDL) induces a reproducible model of biliary cirrhosis in rats, leading to liver failure, portal hypertension, translocation of bacteria and immune system dysfunction. BDL rats show hyperammonemia and decreased motor activities, but only low-grade encephalopathy[45,93-95]. It is possible to reproduce the human neuropathology of type C HE, Alzheimer type II astrocytosis, altered brain osmolytes, low-grade brain edema,inflammation and motor activity deficits by feeding BDL rats with ammonium salts.Of note, by giving ammonium salts to BDL rats, it is possible to obtain a model of acute-on-chronic HE[96].
HE MODELS RESULTING FROM PURE HYPERAMMONEMIA
These models are usually limited to rats and mice and are used to study the effects of hyperammonemia on brain function, in the absence of liver damage or portacaval shunting. They are produced by means of the administration of high-ammonia diets,parenteral infusion of ammonia or urease treatment. These models are inexpensive and easily reproducible and result in animals with impaired memory and learning skills[44]. However, the disadvantages of these models include the time involved to produce alterations and their unsuitability for long-term studies[45].
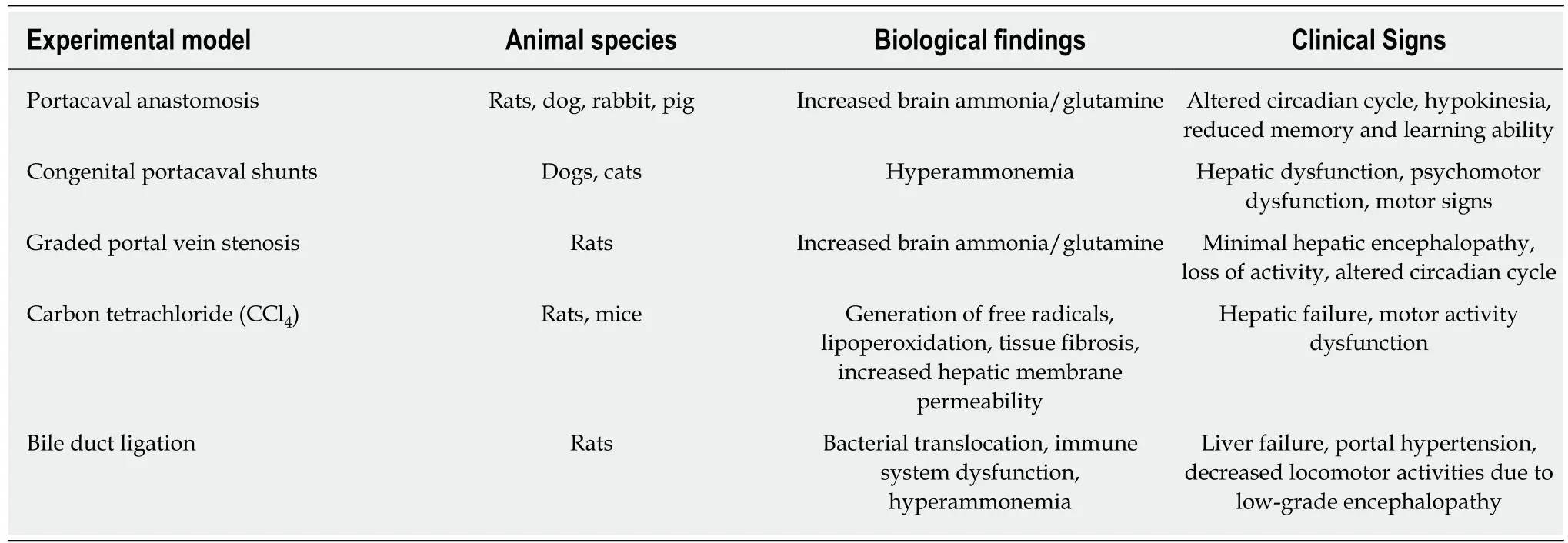
Table 3 Brief description of findings obtained with types B and C experimental models of hepatic encephalopathy
CONCLUSION
HE is a very severe complication in the context of liver failure. HE can be subdivided according to the type of hepatic injury, namely, type A, which results from ALF, type B, which is associated with a portosystemic shunting without intrinsic liver disease,and type C, which is due to chronic liver disease. The pathophysiology of HE has two major factors, namely, increased ammonia levels and neuroinflammation.
Preclinical models have been very useful in investigating the mechanisms of HE and in evaluating novel therapeutic approaches. The most used animal species are rats and mice. Experimental models of ALF (type A) include surgical procedures(hepatectomy and hepatic devascularization) and the administration of hepatotoxic medications. Surgical models resemble postoperative liver failure in humans, whereas pharmacological models are similar to adverse drug reactions due to hepatotoxicity.
Ideally, models of HE associated with portosystemic shunting (type B) and due to chronic liver disease (type C) must exhibit liver cirrhosis, a precipitant factor,neuropathological and neurochemical alterations. These models are generally surgically induced lesions in liver tissue, which evolve to hepatic cirrhosis.
In conclusion, preclinical models have allowed the comprehension of the pathways related to neurological damage as a consequence of acute and chronic liver injury,resulting in the identification of potential therapeutic targets.
杂志排行

World Journal of Hepatology的其它文章
- Bariatric surgery in patients with non-alcoholic fatty liver disease -from pathophysiology to clinical effects
- Comprehensive analysis of HFE gene in hereditary hemochromatosis and in diseases associated with acquired iron overload
- Clinical outcomes after major hepatectomy are acceptable in lowvolume centers in the Caribbean
- Central line-associated bloodstream infection among children with biliary atresia listed for liver transplantation
- Parallel transjugular intrahepatic portosystemic shunt with Viatorr®stents for primary TlPS insufficiency: Case series and review of literature
- Necrolytic acral erythema in a human immunodeficiency virus/hepatitis C virus coinfected patient: A case report