基于环化腺苷的自发荧光特性用荧光光谱法直接测定注射剂的中环磷腺苷
2019-03-11翁文婷谢晓兰黄晓平陈檐芬
翁文婷, 谢晓兰, 黄晓平, 陈檐芬
(1.泉州师范学院化工与材料学院,福建泉州 362000; 2.华侨大学材料科学与工程学院,福建厦门 361021)

图1 环磷腺苷(cAMP)的化学结构式Fig.1 Chemical structures of cAMP
环磷腺苷(cAMP)为6-氨基-9-β-D-呋喃核糖基-9H-嘌呤-4′,5′-环磷酸氢酯,别名环化腺苷酸,其结构见图1。cAMP作为蛋白激酶致活剂,是人体内广泛存在的具有生理活性的一种重要物质,它作为参与调节细胞功能的第二信使物质,在临床上应用广泛。cAMP属于核苷酸的衍生物,是由三磷酸腺苷(ATP)通过腺苷酸环化酶(AC)催化下反应生成,对于多种体内功能活动均起到调节作用[1]。自从1998年发现了cAMP效应分子Epac,使人们对cAMP信号通路中的相关分子与途径有了新的认识[2 - 3]。最近的研究结果显示,受上游信号cAMP刺激时,cAMP直接激活的交换因子Epac会将小分子G蛋白酶Rap结合的无活性(GDP)置换为活性(GTP),并激活Rap,从而使Rap发挥重要信号分子作用,进而调节细胞增殖、基因表达或者细胞黏附和迁移等系统机能活动[4]。这也证明了信号分子cAMP对于调节机体整体活动的协调性和精确性等方面起重要作用[5 - 6]。因此,对cAMP含量的准确检测方法的研究很有意义。cAMP早前运用较广泛的是Gilman的蛋白结合检测法[7]和免疫检测法[8],其他生物荧光方法也能较好追踪活体细胞中cAMP的变化[9],但是操作相对复杂。盛国荣等采用计算分光光度法建立复方cAMP中环乳膏的质量控制标准[10],但需要进行复杂的计算分离过程,线性范围较窄。高效液相色谱(HPLC)作为灵敏的药物分析检测手段被收录在《中国药典》(2015版)中[11],运用该方法对cAMP的药理和含量检测的研究也有报道[12 - 14]。荧光分光光度法因灵敏度高、操作简单、快速灵敏等优点已广泛应用于生物小分子的检测和药理分析的研究中[15 - 16]。基于荧光共振能量转移(FRET)效应,可设计生物荧光传感器用于精确研究活体细胞中cAMP的药理和动态变化[17 - 20]。但是,利用cAMP的自身结构的荧光效应,采用直接荧光光谱法测定其含量的研究罕见报道。
我们前期研究发现,cAMP水溶液具有非环状的腺苷结构不具备的自发荧光性质,而且荧光光谱明显区别于嘌呤环所产生的荧光信号。通过加热并进行溶液条件的优化后,pH=2.0的cAMP溶液在最佳激发波长285 nm条件下,于393 nm处具有最佳的荧光发射信号,荧光量子产率为4.28%,具有稳定的光漂白性和光谱不依赖性质。本实验利用cAMP的含量与荧光强度在一定条件下呈良好的线性关系,建立起一种直接快速的测定环磷腺苷注射剂中cAMP含量的新方法。与现有检测方法相比,该直接荧光法具有准确度高、操作简单、成本低廉和绿色环保等优点。
1 实验部分
1.1 仪器与试剂
Cary eclipse荧光分光光度计(美国,Varian公司);UV-2600紫外-可见分光光度计(日本,Shimazu公司);F-7000荧光分光光度计(日本,Hitachi公司);FLS920荧光光谱仪(英国,Edinburgh Instruments);HPLC-6460Q液-质联用仪(美国,Agilent公司),配电喷雾离子(ESI)源;ZetaPALS Zeta电位及粒径分析仪(美国,布鲁克海文仪器公司)。
腺嘌呤(ANE,Mr=135.13),腺嘌呤核苷(ANS,Mr=267.24),腺苷一磷酸二钠(AMP,Mr=499.19),环磷腺苷(cAMP,Mr=329),均为分析纯试剂,购于上海阿拉丁试剂公司。准确称取ANE、ANS、AMP和cAMP的对照品,分别配制1.0×10-2mol/L的储备溶液,保存于4 ℃冰箱。临用前根据需要,用0.1 mol/L HCl和NaOH溶液将储备液稀释成不同pH的1.0×10-4mol/L工作液。pH=1.98~10.38 B-R缓冲溶液:由0.04 mol/L的H3PO4、H3BO3和HAc混合酸加入不同量的0.2 mol/L NaOH配制而成。其余的试剂均为分析纯;实验所用水为超纯水。
cAMP注射剂(山东潍坊制药厂有限公司,批号:745150501,745150505,745150507):规格为40 mg:5 mL,用超纯水稀释成40 mg/L溶液,置于冰箱中5 ℃储存。
1.2 实验方法
1.2.1荧光光谱和紫外光谱分析移取5 mL对照品操作液,加入等量的不同pH缓冲溶液,稀释成5.0×10-5mol/L溶液,于荧光分光光度计上,设置狭缝5.0/10.0 nm,激发波长λex=285 nm,在290~550 nm范围内进行荧光光谱分析。同时在紫外分光光度计上扫描吸收光谱。设置光谱狭缝为2.5 nm,以纯溶剂为空白参比液,在300~600 nm范围内进行吸收光谱分析。
1.2.2cAMP的高效液相色谱-质谱(HPLC-MS)分析采用ZORBAX Eclipse Plus C18(RRHD)色谱柱(50×2.1 mm,1.8 μm),以超纯水作为流动相A,以色谱纯甲醇作为流动相B,梯度洗脱,流速0.5 mL/min,柱温25 ℃;进样量5 μL。质谱采用ESI离子源;扫描方式:正、负离子同时扫描;采集方式:多反应监测(MRM);离子源温度:350 ℃;毛细管温度:300 ℃;毛细管电压:正、负离子模式下均为4.0 kV;鞘气气流:11 L/min。取cAMP标准品和注射剂各适量,加流动相A制成每1 L中分别含50 μg的操作溶液,作为系统适用性试验溶液,进行高效液相色谱实验。
1.2.3cAMP注射剂样品的荧光光谱测定准确移取cAMP注射液,稀释200倍,制成40 μg/mL溶液后,再稀释成0.4 μg/mL溶液,于荧光分光光度计上,设置狭缝5.0/10.0 nm,激发波长λex=285 nm,在290~550 nm范围内进行荧光光谱分析,同时用水进行空白对照实验。
2 结果与讨论
2.1 cAMP的内源性荧光机理初探
按照操作步骤,分别配制不同pH条件下1.0×10-4mol/L的ANE、ANS、AMP和cAMP溶液,进行荧光光谱测定,如图2所示。在各种溶液体系中,只有ANE因刚性的嘌呤环平面结构而具有自发荧光特性和稳定的荧光强度,荧光光谱的激发峰位置随着pH的增加而发生红移并伴随着Stocks位移的减小(表1),这是因为结构上的伯胺基团质子化程度不同而导致的。在酸性条件下,4种物质均有明显荧光信号,ANE的最大激发和最大发射波长分别为287、381 nm,光谱的Stokes位移是94,当分子上连接上戊糖结构后,无论是核苷还是腺苷结构,荧光光谱的Stokes位移明显增大到105以上,当在ANS的结构上引入磷酸形成AMP,磷酸的位阻效应使杂环结构刚性增强,而且在强酸性环境下易质子化,从而使原来荧光体的最低单线激发态S1为n、π*型转化为π、π*,因此荧光强度明显增强。这也进一步解释了在中性溶液环境中,非质子化的非刚性结构导致ANS和AMP没有荧光特性[21]。

图2 不同pH条件下1.0×10-4 mol/L ANE、ANS、AMP和cAMP的荧光光谱图 Fig.2 Fluorescence spectra of ANE,ANS,AMP and cAMP in different pH with 1.0×10-4 mol/L

表1 ANE、ANS、AMP和cAMP的荧光光谱特征峰数据(nm)
但是一旦磷酸链接到糖环上形成cAMP,两个大杂环结构因C-C键隔开,形成特殊的基态转动构型,导致cAMP在中性环境中也具有特有的自发荧光现象。此时,cAMP的最大激发和发射波长分别为285、393 nm,具有最大的Stokes位移,可明显区别于嘌呤体系。对应的紫外光谱如图3(a)所示,4种物质的吸收峰位置均相同,说明发生紫外吸收的特征结构没有明显改变。很明显采用紫外光谱和紫外检测器的高效液相色谱法来测定cAMP的含量,其方法选择性不如荧光光谱。因此通过测定cAMP的荧光强度进行含量分析具有较高可行性。

图3 B-R缓冲溶液(pH=2.0)中ANE、ANS、AMP与cAMP的紫外(UV)光谱图(a)和cAMP的色谱(b)与质谱图(c) Fig.3 UV spectra of ANE,ANS,AM,cAMP in B-R buffer(pH=2.0)(a),chromatograms(b) and mass spectrum(c) of cAMP
2.2 cAMP的质谱分析
按实验方法中操作步骤,对cAMP进行结构分析,结果如图3(b)所示,在最优化条件下得到的质谱图上有明显三组峰(m/z327.9、m/z133.8、m/z106.9),分别对应着cAMP脱去一个H+的主分子离子峰,C-C 断裂形成的ANE碎片峰和脱去了磷酸后的戊糖分子碎片峰。实验结果表明没有其它杂峰存在,进一步说明cAMP具有稳定的两个杂环结构,可在强酸性条件下以自身特殊的构型存在,并形成自发荧光。
2.3 cAMP荧光体系的条件优化
2.3.1溶剂类型和用量的影响按照操作步骤,分别配制1.0×10-4mol/L cAMP的水、石油醚、正丁醇、乙酸乙酯和甲醇溶液,混匀、静置15 min后。分别在荧光分光光度计上测量荧光光谱。实验结果表明,除在纯水中,其他溶剂均会干扰cAMP的荧光信号,使其荧光峰位置发生改变,强度下降。在纯水环境下cAMP的荧光达到最强,所以本实验选择纯水作为溶剂。
2.3.2缓冲溶液类型和pH值的影响测定了不同pH值条件下cAMP水溶液的荧光光谱,结果如图4所示。酸性条件下cAMP具有较强的荧光信号,荧光强度随溶液酸性增强而逐渐变大,在pH=2.0时荧光强度最大,酸性进一步增强荧光强度反而下降。这可能是因为分子结构中的磷酸根在pH=2.0左右最易质子化而导致的。随后考察pH=2.0的邻苯二甲酸氢钾、NaHPO4-柠檬酸、柠檬酸-NaOH-HCl和B-R缓冲溶液对荧光信号的影响。结果表明,cAMP在B-R缓冲液中荧光最强。所以本实验选择pH=2.0的B-R缓冲溶液为最佳反应条件。
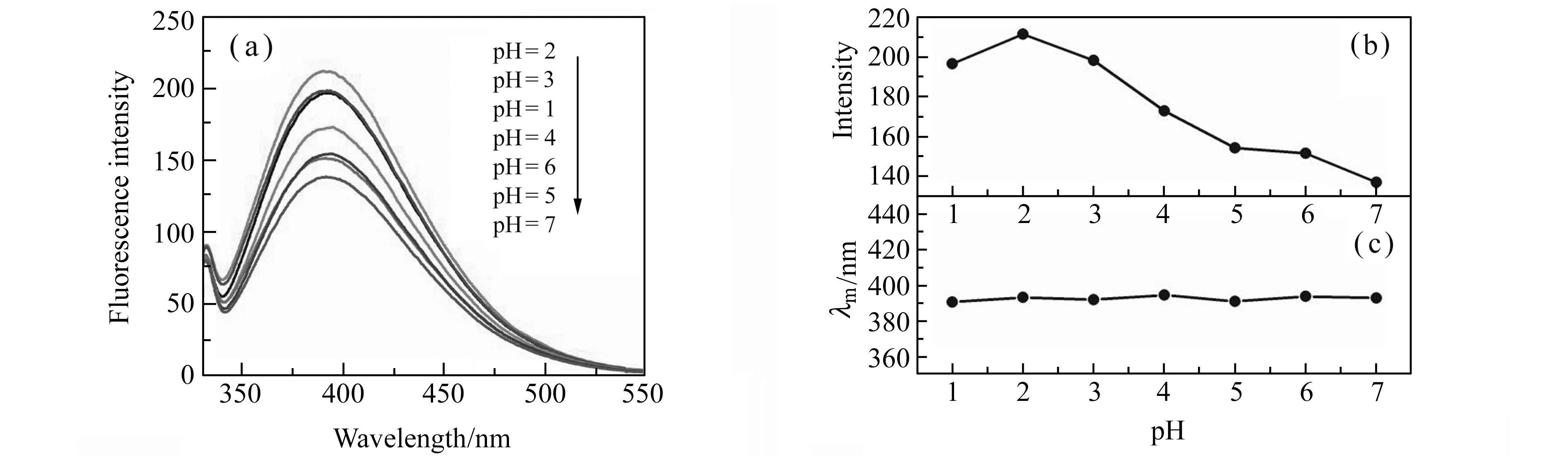
图4 不同pH的B-R缓冲溶液中cAMP的荧光光谱(a)、荧光强度(b)和荧光发射波长(c)随pH的变化趋势图Fig.4 The fluorescence spectra of cAMP at different pH B-R buffer(a),relationship between fluorescentce intensity(b) and emission wavelength(c) with pH

图5 不同激发波长下cAMP的发射光谱(a)、发射波长(b)和荧光强度随激发波长的变化(c)趋势图(狭缝5.0/5.0 nm)Fig.5 The emission spectra of the cAMP solution at different excitation wavelength(a),emission wavelength(b) and relationship of fluorescentce intensity(c) with excitation wavelength(slit=5.0/5.0 nm)
2.3.3温度的影响按照实验方法中的步骤,将配制好的cAMP溶液分别在不同水浴温度下加热2 h,冷却到室温法后,在荧光分光光度计上测定荧光光谱。结果显示,体系在50 ℃温度下反应一定时间,可有效增强其荧光强度,所以本实验选择50 ℃作为最佳加热温度。
2.3.4重现性实验取cAMP标准操作溶液,进行最优化条件处理后,于荧光分光光度计上测定荧光强度[16],平行测定10次,其相对标准偏差(RSD)为1.08%。说明该体系具有较好的重现性。
2.3.5cAMP溶液的荧光光谱性能按实验方法,采用不同激发波长对cAMP进行发射光谱扫描,结果如图5所示。在不同波长激发下cAMP的荧光发射光谱的最大发射峰位置没有明显改变,但是荧光强度先增后降,在激发为285 nm时达到最高。这种激发光谱不依赖性质和荧光染料分子发光的特性是一致的,这也进一步验证cAMP自发荧光的可行性。
2.4 工作曲线与检出限
按照实验方法,准确移取一系列不同浓度的cAMP标准溶液,于荧光分光光度计上测定荧光强度。结果表明:cAMP在1.0×10-6~1.0×10-5mol/L浓度范围与其荧光强度呈现良好线性关系,线性方程为:F=44.87+1.897×107ccAMP(mol/L),相关系数r=0.9991,检出限(3S0/K)为1.708×10-9mol/L。将该荧光法测定所得结果与高效液相色谱法[11]相比较,结果一致。
2.5 共存物质的影响

2.6 注射剂中CAMP的含量测定
取稀释后注射剂样品溶液,分别添加低、中、高3个水平的混合标准溶液,在优化的实验条件下目标化合物的回收率在98.3%~110.0%之间,RSD≤4.9%(表2),可满足实际分析的要求。
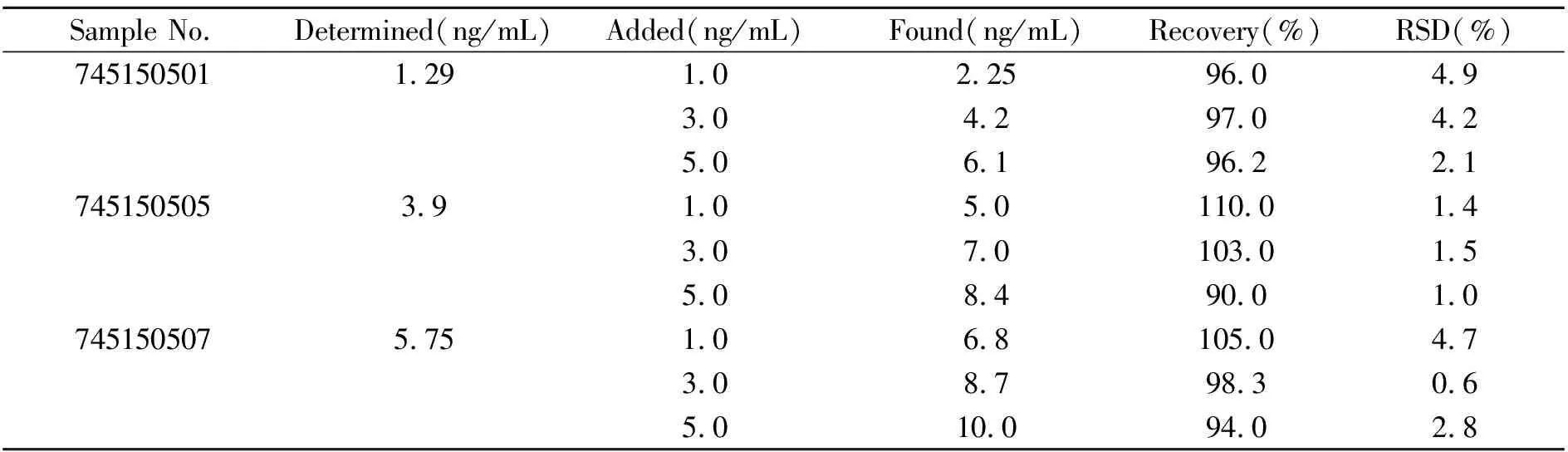
表2 注射剂样品测定结果与加标回收率(n=6)
3 结论
环磷腺苷在酸性至中性溶液环境中会呈现稳定的荧光光谱,最佳激发/发射波长为285/393 nm。在pH=2.0的B-R缓冲溶液中,50 ℃水浴加热2 h后达到最佳荧光强度,量子产率为4.28%。由此建立了一种直接荧光法测定cAMP的新方法。实验表明,本法操作简便,重现性好,精确度高等优点,测定结果较准确,可用于注射剂中环磷腺苷含量的测定。
