绿原酸亲水性磁性分子印迹树脂的合成及其固相萃取性能评价
2019-03-08施树云
彭 胜, 李 奂, 施树云,*
(1. 吉首大学林产化工工程湖南省重点实验室, 湖南 张家界 427000; 2. 吉首大学杜仲综合利用技术国家地方联合工程实验室, 湖南 吉首 416000; 3. 中南大学化学化工学院, 湖南 长沙 410083)
绿原酸(chlorogenic acid, CGA)广泛存在于蕨类植物到高等双子叶植物及相关的中成药中,具有抗氧化、抗菌、抗病毒、消炎、降脂、护肝和神经保护等方面的功效[1]。CGA是众多中药及其制剂的质量控制指标之一,因此建立CGA准确灵敏的定性定量分析方法至关重要。
目前定性及定量分析的常用方法是高效液相色谱法(HPLC),但是考虑到中药及其相关产品成分复杂,在HPLC分析之前有必要对样品进行预处理[2]。固相萃取因高富集因子、操作简便等优势被广泛使用,反相C18、SiO2、金属氧化物、碳基吸附剂和聚合物吸附剂是常用的固相萃取吸附材料[3]。其中,分子印迹聚合物(molecularly imprinted polymers, MIPs)具有构效预定性、特异识别性和广泛实用性等优势,可用于复杂体系中目标组分的高效特异性分离富集,并消除基质干扰[4-8]。迄今,科研工作者在MIPs的合成和应用方面已做出了卓有成效的工作,多数MIPs是在有机溶剂或非极性溶剂中,氮气保护下,通过模板分子与烯丙基功能单体(如甲基丙烯酸、丙烯酰胺、乙烯基咪唑等)及交联剂共聚而成,合成的MIPs在非极性溶剂中具有良好的特异性识别性能[9]。同时表面印迹技术的应用也使印迹位点位于MIPs的表面,增强吸附性能,如已在磁性Fe3O4、中空纤维、纳米TiO2、石墨烯碳纳米管等表面成功地合成了基于CGA特异性吸附的MIPs[10-14]。但该类MIPs在水溶液中非特异性吸附强,而常见的中药制剂、食品、生物制品等均是水溶液体系,因此设计水体系中具特异性识别性能的MIPs势在必行。
本文以CGA为模板、间苯二酚和三聚氰胺为双功能单体、甲醛为交联剂、磁性介孔二氧化硅(Fe3O4@mSiO2)为载体合成了亲水性磁性MIRs(magnetic MIRs, MMIRs)。优化了合成条件,研究了MMIRs的静态、动态和选择性吸附性能,并结合HPLC用于杜仲黑茶中CGA的选择性分离和含量检测。结果表明该方法简便、快速,在水体系中的选择性识别性能好。
1 实验部分
1.1 仪器、试剂与材料
安捷伦1200高效液相色谱系统(在线脱气机、四元泵、恒温柱温箱、自动进样器和二极管阵列检测器) (Agilent Technologies, USA); Nicolet 6700傅里叶红外光谱仪(Thermo Nicolet Co., USA); JEM-2100F透射电子显微镜(JEOL, Japan); JC2000D1接触角测量仪(上海中晨数字技术设备有限公司,上海); VSM7407振动样品磁强计(Lake Shore, USA); SDTQ600热重分析仪(TA, USA); Milli-Q超纯水仪(Bedford, USA)。
三聚氰胺、间苯二酚、甲醛、聚乙二醇6000(PEG 6000)、十六烷基三甲基溴化铵(CTAB)、正硅酸四乙酯(TEOS)、氢氧化钠(NaOH)、三氯化铁(FeCl356H2O)、甲醇和乙醇均购自国药集团化学试剂有限公司(上海); HPLC级甲醇和乙酸购自西格玛奥德里奇贸易有限公司(上海); CGA、咖啡酸(caffeic acid, CA)、阿魏酸(ferulic acid, FA)、肉桂酸(cinnamic acid, CMA)对照品购自西亚试剂有限公司(山东)。
杜仲黑茶由张家界茶坤缘生物科技开发有限公司提供。
1.2 MMIRs的制备
图1为MMIRs的合成示意图。首先合成Fe3O4@mSiO2,步骤同课题组前期研究[7]。接着将间苯二酚(3 mmol)和甲醛(6 mmol)溶于超纯水(30 mL), 40 ℃下磁力搅拌1 h形成A溶液;将三聚氰胺(1 mmol)和甲醛(3 mmol)溶于超纯水(10 mL), 80 ℃下磁力搅拌至透明,冷却至室温形成B溶液。然后将Fe3O4@mSiO2(100 mg)、A溶液、B溶液和CGA溶液(0.04 mol/L, 10 mL)混合,80 ℃恒温机械搅拌反应3 h。磁分离Fe3O4@mSiO2/MIRs后,水洗3次,将其分散于NaOH水溶液(50 g/L, 20 mL)中搅拌除去mSiO2和CGA。最后,磁分离MMIRs,用水洗至中性,40 ℃真空干燥至恒重。
亲水性磁性非分子印迹树脂(magnetic non-molecularly imprinted resins, MNIRs)的制备除了不加模板CGA外,所有步骤同MMIRs的合成。

图 1 MMIRs合成示意图Fig. 1 Synthetic route for preparing MMIRs
1.3 HPLC分析
Waters SunFire-C18色谱柱(250 mm×4.6 mm, 5 μm, Waters);流动相为0.4% (v/v)乙酸(A)和甲醇(B),梯度洗脱:0~10 min, 12%B; 10~16 min, 12%B~18%B; 16~25 min, 18%B;柱温:25 ℃;进样量:20 μL;流速:0.8 mL/min;检测波长:254 nm。
1.4 吸附实验
吸附动力学:准确称取若干份MMIRs(MNIRs) (10.0 mg),分别加入到CGA(1.4 g/L)的50%(v/v,下同)乙醇水溶液(5.0 mL)中,于25 ℃下200 r/min振荡吸附,在不同的时间点(10~100 min)取出,使用HPLC方法检测CGA的质量浓度Ct(g/L)。由方程(1)计算出MMIRs(MNIRs)对CGA的吸附量Qt(mg/g):
在现场环境监测阶段,有很多不确定性,这些问题多多少少会对数据收集的精准度带来影响,要想避免环境监测数据受到破坏,就要对监测人员提出质量控制请求,在采样工作中,要有专业监控人员指导,发现问题及时解决,确保环境监测数据收集行为的规范性。
(1)
式中m(g)表示MMIRs(MNIRs)的质量,C0(g/L)和V(mL)分别是CGA溶液的初始浓度和体积。
静态吸附:准确称取若干份MMIRs(MNIRs) (10.0 mg),分别加入到含不同质量浓度CGA(0.4~1.6 g/L)的50%乙醇溶液(5.0 mL)中,于25 ℃下200 r/min振荡吸附70 min,使用HPLC方法检测CGA的平衡浓度Ce(g/L)。由方程(2)计算出MMIRs(MNIRs)对CGA的平衡吸附量Qe(mg/g):
(2)
选择性吸附:选择模板分子CGA和3个结构类似物CA、FA和CMA,准确称取4份MMIRs(MNIRs) (10.0 mg),分别加入到含CGA、CA、FA和CMA的50%乙醇溶液中(1.4 g/L, 5.0 mL),于25 ℃下200 r/min振荡吸附70 min,使用HPLC方法检测平衡时CGA、CA、FA和CMA的质量浓度,使用方程(2)计算平衡吸附量。
1.5 杜仲黑茶中绿原酸的检测
杜仲黑茶粉碎,称取10.0 g杜仲黑茶粉末于100 ℃下用50%乙醇溶液(80.0 mL)回流提取3次,每次提取2.0 h,合并提取液后抽滤并减压浓缩至干燥,得3.4 g浸膏。将MMIRs (20 mg)加入到杜仲黑茶的50%乙醇溶液中(3.0 g/L, 5 mL),混合物在25 ℃下于200 r/min振荡吸附70 min,磁分离MMIRs,水洗3次除去非特异吸附成分,然后用甲醇-水-醋酸(3∶1∶1, v/v/v, 5 mL)洗脱,并用HPLC分析洗脱液。
2 结果与讨论
2.1 MMIRs的合成和表征
MMIRs的合成过程中采用Fe3O4@mSiO2为载体,CGA、间苯二酚、三聚氰胺和甲醛在mSiO2的介孔中共聚生成MIRs,无需对mSiO2表面进行官能团修饰,最后通过NaOH刻蚀掉mSiO2和模板分子得到MMIRs。N2吸附和解吸附实验显示MMIRs的孔径为2.95 nm,表明MMIRs具有多孔结构、印迹位点位于材料表面。模板分子CGA的加入量及印迹时间对MMIRs的印迹效果影响较大,少量CGA导致识别位点少,然而过量的CGA会和功能单体形成无序自组装;共聚时间的增加会增厚印迹层,从而导致印迹位点的包埋及吸附量的降低。实验中考察了不同CGA的加入量以及不同共聚时间对MMIRs吸附量的影响,最终确定CGA的加入量为0.4 mmol,共聚时间为3 h,合成的MMIRs对CGA具有较好的吸附行为。

图 2 (a)Fe3O4@mSiO2/MIRs和(b)MMIRs的透射电子显微镜(TEM)图Fig. 2 Transmission electron microscopy (TEM) images of (a) Fe3O4@mSiO2/MIRs, and (b) MMIRs
图2为Fe3O4@mSiO2/MIRs和MMIRs的透射电子显微镜(transmission electron microscope, TEM)图,Fe3O4@mSiO2/MIRs的平均粒径约200 nm,其中壳层厚度30 nm左右[7]。除去mSiO2后,MMIRs呈现疏松多孔的核壳结构,外部MIRs层薄约6 nm。Fe3O4、Fe3O4@mSiO2、Fe3O4@mSiO2/MIRs和MMIRs的红外光谱如图3所示,图3a中576 cm-1为Fe-O强的伸缩振动峰,图3b中新增Si-O键在1 089和461 cm-1的振动峰,在2 980 cm-1左右未见明显的C-H吸附峰,表明CTAB的完全除去和Fe3O4@mSiO2的成功合成;图3c中,宽而强的3 405 cm-1来自O-H和N-H的伸缩振动峰,812 cm-1为N-H的面外弯曲振动,1 346 cm-1和1 559 cm-1来自三聚氰胺中C-N的弯曲振动和伸缩振动,1 160 cm-1是C-O-C的伸缩振动;图3d中,未见1 089 cm-1和461 cm-1的吸收峰,显示mSiO2的完全除去。结果表明MMIRs成功合成。

图 3 (a)Fe3O4, (b)Fe3O4@mSiO2, (c)Fe3O4@mSiO2/MIRs和(d)MMIRs的红外光谱图Fig. 3 FT-IR spectra of (a)Fe3O4, (b)Fe3O4@mSiO2, (c)Fe3O4@mSiO2/MIRs, and (d)MMIRs

图 4 MMIRs的(a)热重分析,(b)磁滞回线和(c)水滴的轮廓图Fig. 4 (a) Thermogravimetric analysis, (b) hysteresis loop, and (c) profile of a water drop on MMIRs
热重分析(图4a)显示温度升高到800 ℃时质量损失60%,源于MIRs层的分解;磁滞回线(图4b)显示MMIRs的磁饱和强度为40.6 emu/g,实验中MMIRs在外加磁场的作用下5 s内可实现磁分离,撤除外加磁场后能快速分散,说明制备的MMIRs具有超顺磁性、高磁响应性的特点;水滴与MMIRs接触2 s内迅速铺展,接触角为4.50°(图4c),表明MMIRs具有极强的亲水性。因此,MMIRs可实现水体系中目标化合物的快速选择性分离。
2.2 MMIRs(MNIRs)对CGA的吸附
MMIRs(MNIRs)对CGA的吸附动力学曲线如图5a所示,随着时间的延长吸附量逐渐增大,MMIRs在70 min时对CGA的吸附达到饱和,而MNIRs在40 min时就趋于饱和吸附,MMIRs对CGA的吸附量远远大于MNIRs,主要原因为MMIRs存在CGA的特异性吸附位点,而MNIRs为表面分子间的氢键或静电作用。前期关于MIRs的合成报道中,聚合时间为24 h左右,平衡吸附时间为10~12 h[18-23]。因此,表面多孔MIRs层结构以及适宜的MIRs层厚度可以使印迹位点位于表面,加快吸附速率。
MMIRs(MNIRs)对CGA的静态吸附曲线如图5b所示,随着CGA质量浓度的增加吸附量逐渐增加,在质量浓度大于1.4 g/L时吸附达到平衡,MMIRs对CGA的平衡吸附量(50.87 mg/g)是MNIRs平衡吸附量(9.71 mg/g)的5.24倍,表明了MMIRs对CGA的特异性吸附。迄今,文献报道的基于CGA的MIPs合成均为非水体系,在乙腈等非质子性溶剂中具备选择性吸附性能[10-14]。本文合成了在水体系中具有高特异性吸附的印迹材料。
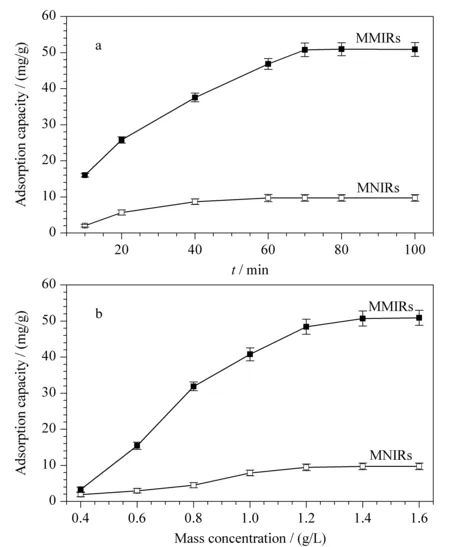
图 5 MMIRs(MNIRs)对CGA的(a)动态吸附和(b)静态吸附等温线(n=3)Fig. 5 (a) Kinetic adsorption and (b) equilibrium adsorption curves of CGA on MMIRs(MNIRs) (n=3)

图 6 MMIRs(MNIRs)对CGA、CA、FA和CMA的吸附容量(n=3)Fig. 6 Adsorption capacities of CGA, CA, FA, and CMA on MMIRs/MNIR (n=3)
MMIRs(MNIRs)对CGA、CA、FA和CMA的平衡吸附结果如图6所示,MMIRs对CGA的吸附容量(50.87 mg/g)是CA(12.05 mg/g)的4.22倍,FA(16.79 mg/g)的3.03倍,CMA(14.18 mg/g)的3.59倍;CGA、CA、FA和CMA在MNIRs上的吸附容量差异不大。结构的差异性会影响吸附能力,说明印迹孔穴与CGA的互补性。
MMIRs对CGA吸附的稳定性至关重要,合成了5批MMIRs,其吸附量的RSD值为6.4%,说明了批间的稳定性;考察了10次连续吸附-解吸附的实验结果,随着循环次数的增加,MMIRs对CGA的吸附能力轻微下降,10次循环后吸附量为原先的91.5%,表明MMIRs对CGA吸附的稳定性和重复利用性能。
2.3 MMIRs-HPLC方法的建立
配制系列CGA标准溶液,使用MMIRs进行吸附-解吸附实验,HPLC分析检测CGA,在2.0~200.0 mg/L范围内得到CGA的线性方程为Y=52.06X-19.92(R2=0.999 3),其中Y和X分别是CGA的HPLC峰面积和质量浓度(mg/L)。CGA的检出限(信噪比S/N=3)为0.7 mg/L,定量限(信噪比S/N=10)为1.6 mg/L。方法的加标回收率为93.1%~109.4%,相对标准偏差小于9.6%。说明该方法可满足CGA的测定要求。
2.4 实际样品检测
将MMIRs-HPLC方法用于杜仲黑茶中CGA的测定,图7为杜仲黑茶经过MMIRs固相萃取前后的HPLC谱图,经过MMIRs选择性固相萃取后,基本不存在其他干扰性物质。最终,测得杜仲黑茶中CGA的含量为(2.79±0.18) mg/g,结果与文献[14]报道一致,表明MMIRs选择性萃取分离结合HPLC分析检测可用于实际复杂体系中CGA的特异性分析。
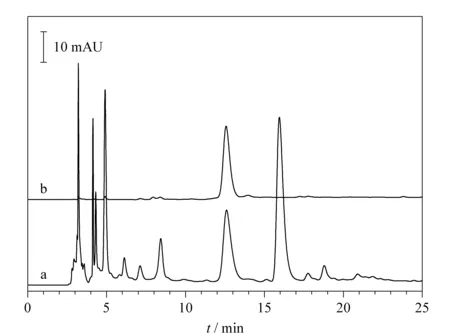
图 7 杜仲黑茶经MMIRs萃取(a)前、(b)后的HPLC谱图Fig. 7 HPLC chromatograms of Duzhong brick tea (a) before and (b) after extraction with MPMIPs
3 结论
本文以CGA为模板、间苯二酚和三聚氰胺为双功能单体、甲醛为交联剂、磁性介孔二氧化硅为载体合成了亲水性MMIRs。mSiO2的多孔结构提供了MIRs的聚合场所和多孔结构,简化合成过程、增加吸附容量、加快吸附速率。MMIRs对CGA表现出良好的吸附选择性,结合HPLC分析用于杜仲黑茶中CGA的测定,排除复杂基质的干扰,提高分析的准确度。MMIRs在水体系中的特异性吸附性能使其可作为新型吸附材料用于生物、环境和临床领域的研究中。
