基于近红外光谱和梯度提升决策树建立当归药材及伪品的定性判别模型*
2019-03-06拱健婷邹慧琴王大仟刘长利
拱健婷,李 莉,邹慧琴,徐 东,王大仟,丛 悦,刘长利
(1.北京市卫生局临床药学研究所 北京 100035;2.首都医科大学附属北京中医医院 北京 100010;3.北京中医药大学中药学院 北京 102488;4.首都医科大学中医药学院 北京 100069)
当归为伞形科植物当归Angelica sinensis(Oliv.)Diels.的干燥根[1],始载于《神农本草经》[2],为常用的名贵大宗药材,素有“十药九归”之称,临床上多用于治疗血瘀证、血虚证,为“血中之圣药”[3]。当归是卫计委公示的“药食两用”品种之一[4],也是保健品、化妆品、饮品、香料的原料,市场需求量大,是产销量位居第二的大宗中药商品[5]。随着其需求的增长和价格的提高,当归混伪品也日渐增多,近年来笔者发现市场上部分药材因名称与当归相似而混为当归药用,如云南野当归(Angelica sp.)、欧当归(Levi sticum officinaleKoch.)和华中前胡(Peucedanum medicum)混作当归。这些植物在根茎形态上与正品当归十分类似,但在药效方面相差甚远[6,7],严重影响用药安全,因此快速有效鉴别当归真伪成为当务之急。
已有许多报道采用性状鉴别法、紫外光谱法、薄层色谱法、分子技术等对当归进行鉴别[7-10],取得了一定的成效,但是仍存在易受人为主观因素影响、样品制备复杂、特征信息较少、结果难量化、成本高、溶剂污染等局限。近红外光谱(Near Infrared Spectrum;NIRS)技术是正在迅速发展的一种绿色分析技术,具有快速、价廉、无损等特点,与化学计量学结合,广泛用于农业、食品、化学和石油化工、制药等领域的定性和定量分析[11],被美、欧、日、韩、澳大利亚等国家药典纳入附录内容[12]。近红外光谱既能全面地反映中药的整体信息,在苦参[13]、大黄[14]、板蓝根[15]、三七[16]等中药真伪鉴别中近红外技术已经得到了很好的应用。此外,近红外技术结合化学计量方法PCA[12,13]、偏最小二乘判别分析[13,17]、ANN[14]、SVM[15]等能够实现当归产地、产期[18]及不同部位[17]的精细鉴别。本研究将NIRS 技术与GBDT 结合应用于当归药材真伪的快速鉴别,旨在建立一种快速、准确、便捷的当归真伪判别模型,规范当归药材市场。
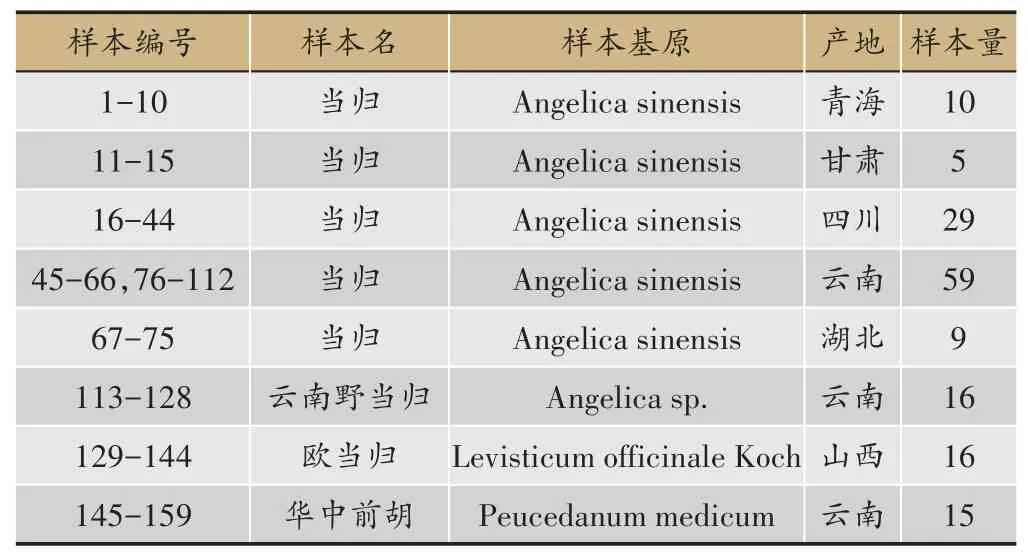
表1 样品信息表
1 材料与方法
1.1 样品来源
本试验中的样品分别于2017年10月至2017年12月期间采集,其中来自不同产地的正品当归5 批次共112份,伪品当归云南野当归、欧当归、华中前胡各1批次,共计47份。
全部样品均由北京市临床药学研究所李莉研究员鉴定,其编号、基原植物、产地信息如表1所示,样品均保存于北京市临床药学研究所202实验室。
1.2 仪器与方法
1.2.1 仪器
NIR-M-R2 型近红外光谱仪(扬光绿能),配备InGaAs 检测器光谱采集范围900-1 700 nm,共228 个变量,采用Hadamard 模式进行扫描。
1.2.2 光谱采集
样品从头部1 cm 处进行切割获得断面,将光纤探头垂直于样品头部断面获取近红外光谱信息。为减小实验误差,每个样品测定3 次,将各次测定得到的光谱曲线加和取平均得到各样品的数据曲线用于后续数据分析。
1.2.3 方法学考察
按照样品测定方法操作,在同一背景下对同一样品进行6 次扫描,求得228 个波长下吸光度的标准偏差为0.000 56-0.004 3,光谱的均方差0.000 051-0.003 9,仪器稳定性良好。
取同一份样品,按照样品测定方法操作,采集其切割0、1、2、3、4、5、6 h 后样品断面的光谱数据,求得不同吸收波长下RSD 为0.007 9-0.87%,样品在6 h 内相对稳定。
1.3 数据处理
1.3.1 预处理
由于仪器首尾噪声较大使得光谱前端和后端有较明显的噪声,为避免低信噪比对分析的影响,本实验截取961-1 655 nm 波长范围内的光谱数据作为下一步分析的数据,以初步去除两端噪声干扰。
1.3.2 软件
本研究中PCA 在PAST 3.0 软件上完成,其余算法GBDT、SVM、ANN、随机森林(Random forest;RF)均由Python 2.7实现。
2 结果与分析
2.1 当归及伪品的近红外光谱曲线
本试验采集到的近红外光谱数据如图1 所示,整体来看,当归及伪品的NIRS 曲线变化趋势和特征吸收基本一致,无法直接鉴别当归真伪。从平均光谱图(图1b)可以看出,当归与华中前胡、欧当归、云南野当归样品的光谱曲线吸收率存在区别,平均吸收率大小依次为华中前胡>云南野当归>欧当归>当归,这一差异为当归真伪的鉴别奠定了数据基础。但是由于仪器测量的波长范围局限,更多地得到的是芳烃、甲基、亚甲基、次甲基、水、胺等的合频和倍频吸收峰,信号强度低且峰谱宽,同一波段是样品多种信息的叠加,谱峰重叠严重使得对当归及伪品的光谱进行直接分辨较为困难,因此有必要借助化学计量学算法做进一步分析。
2.2 PCA定性分析
961-1 655 nm 间有198 个变量,数据量大且相邻波段之间的相关性强,造成信息的冗余,选用适当的方法剔除不相关变量十分必要。PCA 目的是降维,消除相互重叠的信息部分,实现用少数关键变量代替全光谱,降低模型运算量和复杂度、提高模型稳定性和预测准确性[19]。PCA 通过提取198 个指标相关矩阵内部相关信息,剔除原始数据中高度冗余的变量,使数目较少的新变量成为原变量的线性组合,而且新变量能最大限度的表征原变量的数据结构特征[20]。
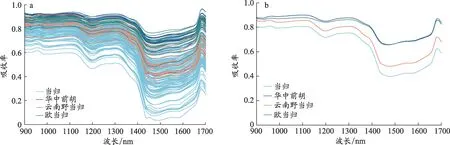
图1 当归、华中前胡、欧当归、云南野当归的近红外原始光谱(1a)和平均光谱(1b)
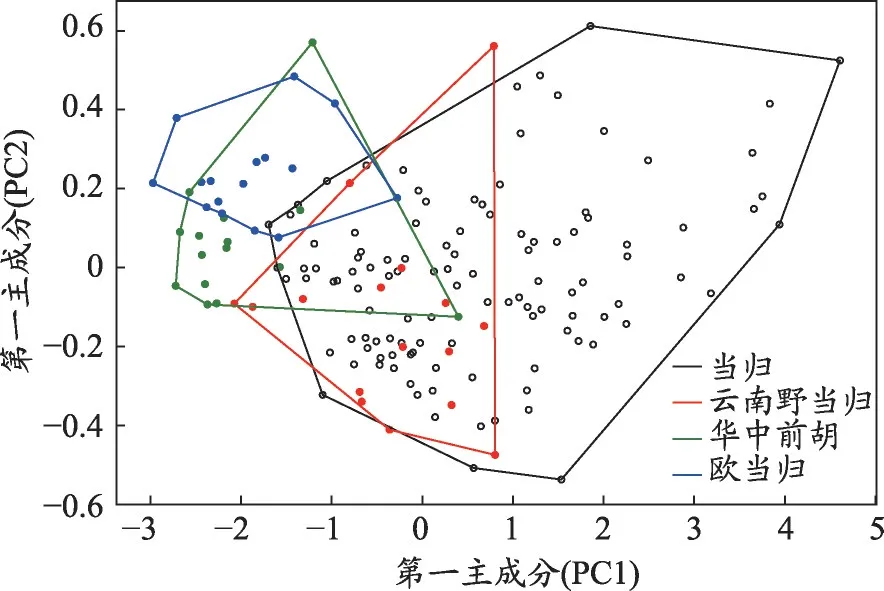
图2 当归、华中前胡、欧当归、云南野当归主成分分析得分图
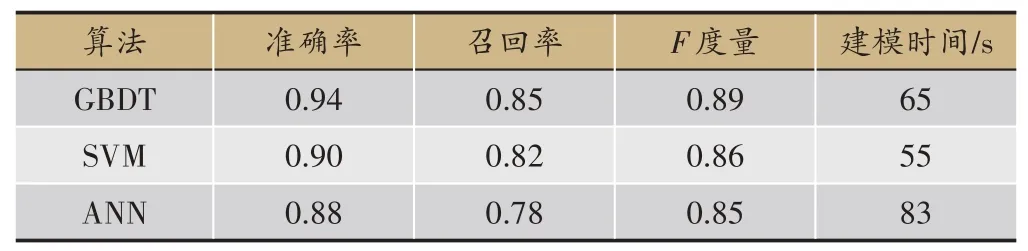
表2 三种算法的性能比较
对所有样品的光谱数据进行主成分分析可知,第一主成分(Principle Component 1,PC1)和第二主成分(PC2)的贡献率分别为97.78%、1.99%,表明PC1 和PC2 已能够表达99%以上的原始光谱信息。由图2可知,当归与华中前胡、欧当归、云南野当归主成分二维投影图中存在交叉无法有效分类,这是因为当归与伪品均为伞形科植物,化学成分较相似,其中当归与云南野当归重叠部分最多,与二者同属当归属亲缘关系较华中前胡、欧当归近有关。若要进一步准确鉴别当归及伪品需对光谱数据进行进一步处理。
2.3 建立GBDT判别模型
2.3.1 GBDT、SVM、ANN模型比较
GBDT 算法由Jerome Friedman[21,22]于2001 年提出,可用于分类和回归,通过集成多个弱学习器CART回归树最终组合成一个强学习器。GBDT 每一次迭代是为了减少上一个模型的拟合残差,并在残差减少的梯度方向上建立新的CART回归树。本文所建判别模型基于Python-Sklearn 工具包实现,实验中所用的计算机配置为Intel Core-i3处理器,2.2GHz主频,4 GB内存。GBDT 参数采用网格搜索方法最终寻找到的最优参数为:树数量为1500、学习率0.01、最大深度为6、一阶正则项系数为0.3、二阶正则项系数为0.4、损失函数为交叉熵损失函数。
为验证提出的分类模型的优越性,将该模型与SVM、ANN 构建的模型进行比较。同样采用网格搜索方法寻找最优参数,最终SVM 分类模型参数为:核函数为RBF(径向基核函数),核函数系数为0.1,惩罚项系数C 为100,最大迭代次数为120 次;ANN 分类模型参数为:输入层神经元228 个、隐藏层神经元10 个、输出层神经元4 个、学习率0.4、激活函数为sigmoid函数。
将正品当归作为正类,其它为负类,分类器在测试数据集上的预测或正确或错误,设置4 种情况出现的样本数量如下:TP 将正类预测为正类的数量,FN 为将正类预测为负类的数量,FP为将负类预测为正类的数量,TN 为将负类预测为负类的数量。这四个量可以导出几个重要的量化评估指标——准确率、召回率以及F度量,用于评价分类算法的性能。

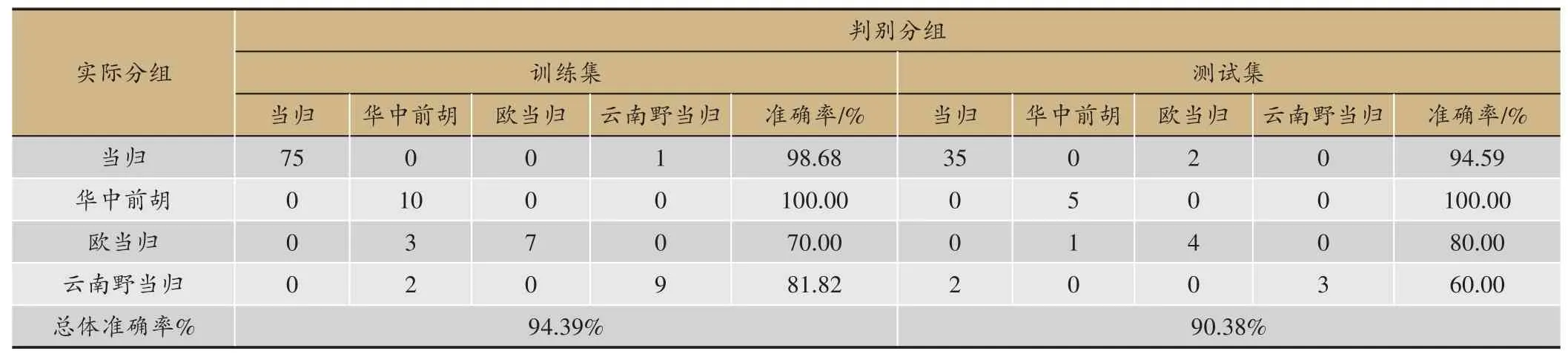
表3 GBDT分类模型准确率
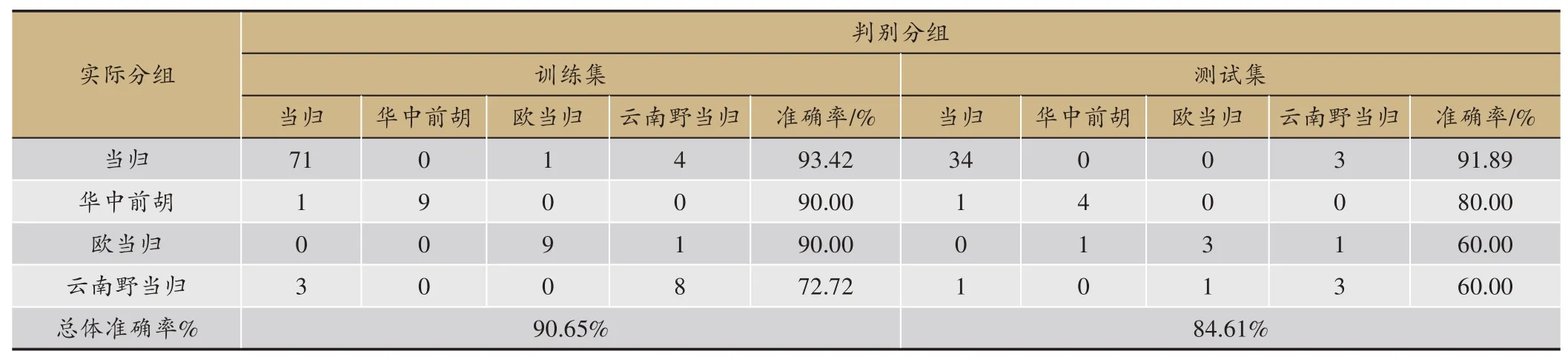
表4 SVM分类模型准确率
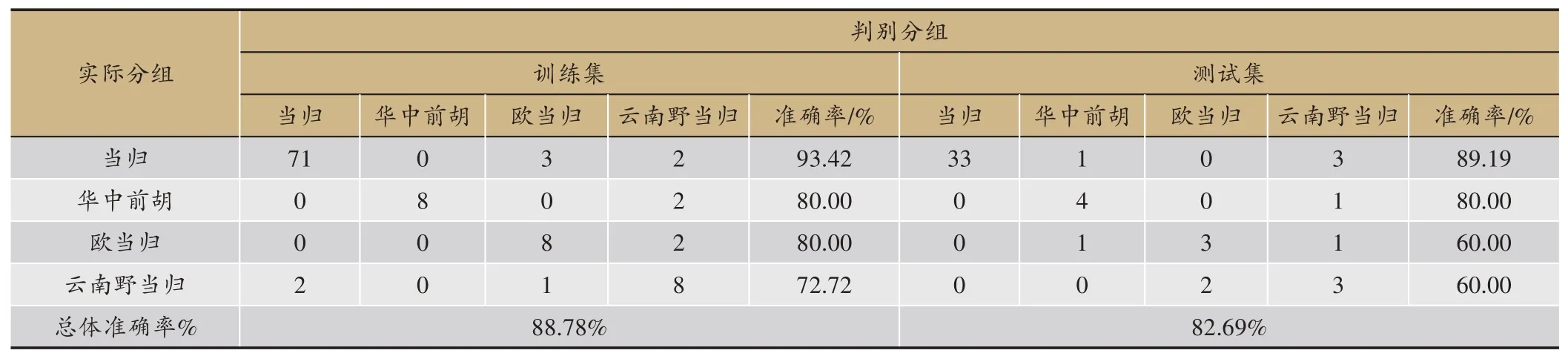
表5 ANN分类模型准确率
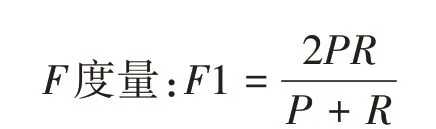
3种算法的预测结果见表2,梯度提升决策树无论是精确度、准确率、召回率还是F值都比另外两种算法要好,由于需要迭代生成很多棵树,所以训练模型的时间略长于SVM;SVM 效果次之,并且在训练模型的过程中只需要寻找惩罚项系数、核函数类型、核函数系数、迭代次数等几个参数,所以耗时比另外两个模型要短;ANN 得到的效果最差,这是因为神经网络模型需要大量的数据样本做支撑,从而用来训练模型参数、学习各个特征之间的相关关系,而本课题的样本量较小,所以导致训练效果最差,并且ANN 需要寻找的网络参数量比较大,导致耗时最长。
随机抽取76 份当归、10 份华中前胡、10 份欧当归、11 份云南野归作为训练集建立判别模型,剩余的37 份当归、5 份华中前胡、5 份欧当归、5 份云南野归作为测试集以评估所建模型的性能,GBDT 鉴别结果如表3 所示,所建立的分类模型对当归的真伪有较好的鉴别效果。训练集与测试集总体判别率分别为94.39%、90.38%,其中GBDT 模型对华中前胡的识别率达到100%。GBDT 识别模式下当归部分样品发生误判,但单组判别准确率也大于90%,可见利用GBDT建立的分类模型能够有效鉴别当归与其混伪品。
2.3.2 RF优化模型
特征选择是指从原始特征集中选择使某种评估标准最优的特征子集,以使在该最优特征子集上所构建的分类或回归模型达到与特征选择前近似甚至更好的预测精度,同时筛除低信息量的冗余特征,达到降低训练模型所需时间、增强模型鲁棒性的目的。RF是Leo Breiman 于2001 年将Bagging 集成学习理论与随机子空间方法相结合而提出的一种机器学习算法[23-25]。RF 具有准确度高、学习速度快、对噪声和异常值有较好的容忍性,对高维数据分类问题具有良好的可扩展性和并行性[26]。它集成多棵决策树的预测,在决策树构建过程中,树的每个结点都是以一定原则度量变量重要性,这一过程实际上就是一个特征选择过程[27]。
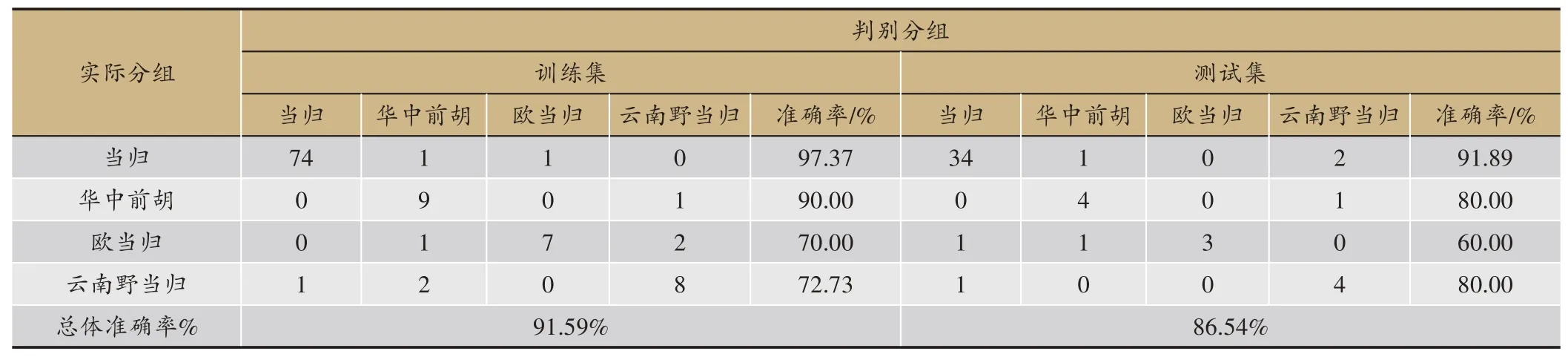
表6 特征光谱判别模型准确率
采用RF 来度量各个特征波长的重要性,步骤如下:①从159 个样本中随机有放回抽取N(本文设置为全量样本的70%,即112 个)个样本,并且随机从198个特征波长中随机选择M(本文设置为总特征的40%,即79 个)个特征波长,构成一个样本子集。重复此过程100 次,得到100 个样本子集;②对100 个样本子集单独训练决策树模型,设置每棵决策树深度为6,不做任何剪枝操作,按照Gini 指数最小原则进行特征分裂,直到该节点下的所有样本都属于同一类或者达到设置的最大深度;③将生成的100 个决策树组成随机森林,按照多棵树分类器投票决定最终的分类结果。同时,统计生成每棵树时所使用的特征波长频次,累加求取均值后得到每个特征波长使用的频次,按照使用频次对198个特征进行从大到小排序。
最终,选择前20个频次高的特征波长作为最重要的特征子集,它们分别是:976 nm、1 016 nm、1 492 nm、1 511 nm、1 521 nm、1 528 nm、1 550 nm、1 573 nm、1 576 nm、1 580 nm、1 586 nm、1 598 nm、1 611 nm、1 621 nm、1 624 nm、1 636 nm、1 640 nm、1 646 nm、1 649 nm、1 655 nm。
近红外光谱振动倍频区有丰富的基团结构信息,一些含有C-H、N-H、O-H 和S-H 化学键的化合物会产生吸收,除在1 400 nm-1 800 nm 之间产生一级倍频,往往还会分别在900 nm-1 200 nm 和780 nm-900 nm 谱带内产生二级倍频和三级倍频,反映的是中药化学成分的综合信息[28]。如本文中970 nm和1 450 nm附近的吸收峰主要是由于样本细胞中水对光谱吸收引起的,分别为O-H 伸缩振动的二级倍频和一级倍频;在1 200 nm 附近的吸收峰与N-H 键有关。RF 筛选出的特征波长均处于一级倍频、二级倍频区,且从图1 可以看出,1 400 nm-1 655 nm 范围内当归的吸光度与华中前胡、欧当归、云南野当归吸光度的差异较大有利于当归真伪的鉴别,因此将RF 所筛选的20 个特征波长用于建立特征光谱判别模型。
2.3.3 近红外特征光谱判别模型的建立
为了建立基于近红外特征光谱的当归真伪判别模型,将所分析出的20 个特征波长作为GBDT 的输入,所得模型的判别效果见表6。相比于原始光谱,特征光谱判别模型所用到的光谱变量大大减少,建模过程得到了简化,特征光谱所建模型判别准确率虽有所下降,但训练集与预测集的正确率仍均高于85%。对4 类样本进行分析,当归单组训练集和测试集判别准确率分别达到了97.37%和91.89%,因此所建立的特征光谱判别模型也能够较好地实现当归的真伪鉴别。
3 讨论
在当归真伪判别研究中发现,PCA 判别分析时区分效果不佳,这表明传统的线性模式识别方法PCA 难以满足鉴别准确性的要求,需要采用更先进的模式识别相关理论和算法来提高近红外光谱技术的识别能力。本文采用GBDT、SVM、ANN 三种非线性方法进行建模分析,识别准确率在训练集和测试集上均大于80%优于PCA。本研究结果显示近红外光谱技术能有效地识别当归及伪品光谱特征差异,并结合GBDT、SVM、ANN 模式识别理论建立了判别模型,为当归及伪品鉴别提供了一种准确而快速的新方法。
近红外光谱虽然信息量大,但由于当归及其混伪品为近缘植物具多种相同成分,使得NIR 光谱图非常相似,不能简单以峰位、峰形进行直接分类,选择合适的数据处理方法提取到特征信息也是分类鉴别的关键。相较于NIR 分析常用的建模方法ANN 和SVM,本文尝试引入一种基于多特征GBDT 的分类方法,利用RF 筛选变量并调整参数训练出最佳预测模型。通过3 种模型判别结果的对比,可以看出,GBDT 模型性能优良,具有较高的预测准确率和很好的适用性,可应用于当归的定性判别分析。然而,针对当归伪品,所建立的判别模型存在误判现象,分析其原因可能是由于伪品较难收集,本研究建立模型的伪品数量有限。有待于在今后实践中扩大校正集和预测集样本容量,完善数据库以优化模型。
本文旨在建立一个快速、简便、无损的当归定性判别模型,以药材断面进行光谱的采集较选择粉末简便、耗时短。目前已有文献报道采集枸杞子表面近红外漫反射光谱实现产地快速识别,基于主根横断面近红外光谱实现西洋参和人参的快速筛查[29,30],以上证明采集药材的断面、表面光谱进行定性鉴别是可行的。此外,市场上存在一些贵重药材掺伪现象,如冬虫夏草、野山参、鹿茸、西洋参、川贝等,此类药材价格昂贵,充分发挥NIR 非破坏性的优势进行直接鉴别研究具有现实意义。我国幅员辽阔,中药品种繁多,“同名异物”和“同物异名”的现象依然存在,即使在今天,中药品种混淆的问题亦有出现,如“关木通”导致马兜铃酸肾病事件[31],香港“白英”和“寻骨风”混淆导致病人患上肾衰竭和尿道癌[32]。如何运用现代科学的理论知识和技术方法来快速、简便、准确地鉴定中药品种,保证临床疗效,是一个迫切的课题。近红外光谱技术能够提中药品种识别的速度和识别能力,满足基层现场快速鉴别的需要。充分发挥近红外自身优势,通过对中药材的大样本量分析,建立稳健的近红外模型,结合云计算和互联网等现代手段,以在全国范围内建立近红外中药品种识别模型网络系统,应用于产地、加工炮制、运输、储存、流通各个环节,从而解决目前存在的品种混乱问题,对中药规范化管理具有重要意义。中药鉴定是一门与时俱进的学问,应在传统经验鉴别的基础上引入现代科学的理论知识和技术方法使中药鉴定更为快捷、科学,推动我国中药现代化进程。
