不同RuO2含量对RuO2-Fe2O3催化剂氨选择性催化氧化性能的影响
2019-03-06,,,,,,
, , , , , ,
(昆明理工大学 环境科学与工程学院, 云南 昆明 650500)
氨气(NH3)作为主要的工业污染气体之一,已经造成一系列严重的环境问题。此外,来自于柴油车尾气后处理系统中的氨溢流是氨气的另一主要来源[1,2]。当人体吸入过多NH3后会造成人体的肺部疾病甚至死亡,所以NH3的有效去除是一个亟待解决的问题。与传统的吸附、吸收和分解法相比,氨选择性催化氧化(NH3-SCO)法作为一类更加有效和环保的NH3处理方法在近些年受到广泛关注[3-5]。其主要反应如下:
4NH3+ 3O2→ 2N2+ 6H2O
(1)
在这一反应过程中,催化材料的选择尤为重要。在过去的研究中,应用于氨催化氧化反应的催化剂主要包括贵金属基催化剂、过渡金属氧化物催化剂和分子筛类催化剂。其中,贵金属Pt、Rh、Pd、Ir和Ag基催化剂具有较好的低温NH3催化活性,但是这些催化剂普遍具有较低的N2选择性[6-9]。过渡金属氧化物CuO、CoOx和MnOx具有较高的氨催化氧化活性,但其N2选择性和贵金属一样低[10-12]。此外,对于贵金属和过渡金属改性的分子筛类催化剂(Pd-Y、Rh-ZSM-5和Cu-ZSM-5)在较宽的温度窗口内具有很高的N2选择性,然而,其反应条件比较苛刻,一般情况下NH3在很高的反应温度下才能达到完全转化[13-15]。
最近几年,氧化铁(Fe2O3)作为一类有潜力的过渡金属氧化物在NO还原[16]、甲苯氧化[17]、CO氧化[18]等催化领域得到广泛应用,其在氨催化氧化领域也得到认可[19,20]。此外,贵金属钌由于其优异的氧化性能和相对较低的成本性在氨氧化领域也得到应用[21,22]。Cui等[21,22]发现,当RuO2的负载量分别达到5%-30%和70%-95%时,RuO2-CuO/Al-ZrO2和CuO/RuO2催化剂表现出较出色的低温催化活性,其在200 ℃左右实现了NH3的完全转化。
考虑到催化材料制备的经济性,本研究制备了一系列具有较低RuO2含量的RuO2-Fe2O3复合氧化物催化剂并将其应用于NH3-SCO反应。本实验主要研究了不同RuO2含量对催化剂催化活性的影响。此外,本研究通过一系列表征方法探究了RuO2含量对催化剂结构特性的影响。原位漫反射红外光谱实验用于催化剂表面酸性位点的研究以及表面催化反应过程的推测。
1 实验部分
1.1 催化剂的制备
采用溶胶-凝胶法制备RuO2-Fe2O3复合氧化物催化剂,所用药品均为分析纯(AR)。首先,将九水硝酸铁(Fe(NO3)3·9H2O)和三水氯化钌(RuCl3·3H2O)溶于60 mL去离子水中,室温搅拌一定时间至完全溶解。然后,将柠檬酸(C6H8O7·H2O)以两倍金属离子总摩尔数的量(n(柠檬酸)/n(Fe3++Ru3+)=2)加入上述溶液中持续搅拌一段时间形成均匀的混合溶液。接下来,将混合液于80 ℃水浴加热直至形成湿凝胶后再放于80 ℃烘箱中干燥72 h。最后,将形成的干凝胶放置于管式炉中氮气氛围下550 ℃焙烧3 h后,再在空气氛围下500 ℃焙烧4 h得到所需的RuO2-Fe2O3复合氧化物催化剂。制备得到的催化剂以RuO2含量不同将其标记为xRuO2-Fe2O3(x为RuO2的质量百分含量,x=0.5%、1%、1.5%、2%)。此外,作为对比,本研究利用相同的方法制备了纯的Fe2O3样品。
1.2 催化剂的表征
X射线衍射(XRD)表征测试采用Bruker D8型X射线衍射仪。测试条件为,CuKα为衍射源(λ=0.15406 nm),所需管电压和管电流分别40 kV和40 mA。20°-85°扫描,扫描速率6(°)/min,步长0.02°。
比表面积(BET)、孔径和孔容是在TriStar II 3020型设备上测定的。在测试前,样品需在300 ℃真空条件下脱气处理4 h。之后以高纯氮为吸附气体进行N2吸脱附处理,以Brunauer-Emmett-Teller (BET)方程计算催化剂的比表面积。
氢气程序升温还原(H2-TPR)测试实验用TCD测定。首先将30 mg样品装于内径为4 mm的石英管中,在300 ℃用高纯氮吹扫40 min,以去除样品表面杂质。之后,冷却至所需温度后,将气路切换为5% H2/Ar混合气,以8 ℃/min的升温速率从50 ℃升温至800 ℃,并以TCD检测其H2消耗量。
NH3程序升温脱附(NH3-TPD)实验是在装配有U型石英管的固定床反应装置中进行的。在吸附NH3前,每个样品都在400 ℃用N2进行1 h的预处理。冷却至室温后开始吸附40 min的NH3,然后N2吹扫20 min。最后N2氛围下进行程序升温脱附实验,升温速率为8 ℃/min。与此同时,NH3的脱附量通过四级杆质谱仪进行实时监测。
原位漫反射红外光谱(In situ DRIFTS)实验是在Nicolet 6700型傅里叶变换红外光谱仪上测试的。首先将样品装于原位红外池内,N2氛围下400 ℃预处理20 min除去表面杂质。将样品冷却至室温后,根据所需测试条件进行条件设定,采用OMNIC软件采集光谱,扣除背景后的谱图即为所需谱图。测试条件:先吸附NH3,NH3体积分数为0.06%,N2做平衡气,总流量为100 mL/min。之后关掉NH3,在N2氛围250 ℃条件下采集不同时间点的红外光谱。
1.3 催化剂的活性评价
样品的NH3-SCO活性评价采用程序升温法,将0.4 mL粒径为40-60目的催化剂装在内径为6 mm的石英管固定床微型反应器中进行程序升温测定,测试温度为150-350 ℃。混合气组分(体积分数)为:0.08% NH3,5% O2,Ar作为平衡气,配气总流量为400 mL/min。分别采用GXH-1050E型氨气分析仪和ECOM·J2KN型烟气分析仪检测反应气尾气中NH3和NOx(NO、NO2)含量,N2O采用电子捕获检测器(ECD)检测。NH3转化率和N2选择性分别通过以下公式计算得出:

(2)

(3)

(4)
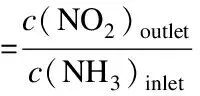
(5)
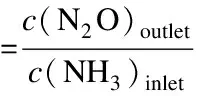
(6)
2 结果与讨论
2.1 催化剂的NH3-SCO活性
图1为纯的Fe2O3和RuO2-Fe2O3催化剂的NH3-SCO催化性能评价结果。
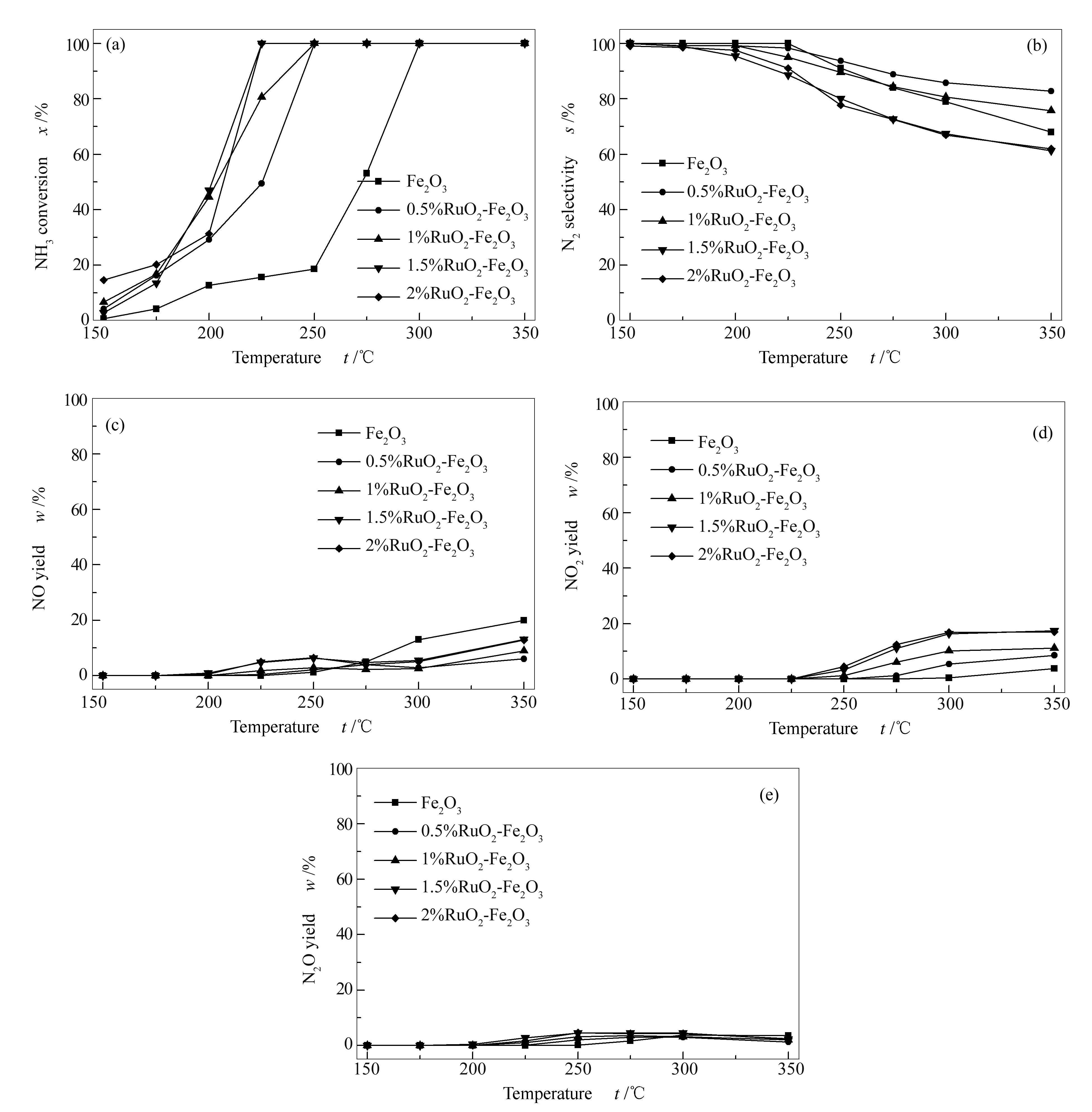
图 1 RuO2含量对RuO2-Fe2O3 催化剂SCO催化性能的影响
由图1可知,Fe2O3表现出较温和的催化活性,在300 ℃左右NH3转化率达到100%,此时N2选择性达到79%。值得注意的是, 随着RuO2的加入,催化剂的催化活性明显提高,表现出较好的低温活性,并且随着RuO2含量的增加活性增加更加明显。当RuO2含量为0.5%和1%时,NH3完全转化的温度为250 ℃,N2选择性在350 ℃时仍大于80%(图1(a)和图1(b))。当RuO2含量为1.5%时,催化剂在225 ℃左右实现100%的NH3转化率和89%的N2选择性。然而,当进一步增加RuO2含量至2%时,催化剂的催化活性并没有增加,反而有下降的趋势,N2选择性没有明显变化。总的来说,RuO2-Fe2O3催化剂的催化活性随着RuO2含量的增加而增加,RuO2含量增加到一定量后反而降低了催化活性, RuO2含量为1.5%催化活性最好。然而,N2选择性的变化趋势则与NH3转化率相反,但其都表现出优异的低温活性和N2选择性。
与此同时,在产物的检测过程中发现存在少量的副产物NO(图1(c))、NO2(图1(d))和N2O(图1(e))。对于纯的Fe2O3样品来说,在反应温度高于250 ℃时才检测到主要的副产物NO和N2O,且只有极少量NO2生成。与之相比,从RuO2-Fe2O3催化剂的各个副产物的产率图中可以看出,在测试温度低于250 ℃时只有极少量的副产物产生,且主要是N2O,这一结果与低温下催化剂的高N2选择性一致。当反应温度高于250 ℃时,N2O产率有所降低,此时的主要副产物是NO和NO2。总的来说,Ru的加入改变了副产物的生成趋势。
2.2 XRD分析
XRD分析揭示了催化剂的结构特征,图2(a)为纯的Fe2O3和RuO2-Fe2O3催化剂的XRD谱图。由图2可知,对于纯的Fe2O3样品,在24.3°、33.3°、35.7°、41.0°、49.6°、54.2°、62.6°和64.1°等位置出现了不同强度的衍射峰,这些衍射峰分别对应于Fe2O3的(012)、(104)、(110)、(113)、(024)、(116)、(214)和(300)晶面,属于典型的赤铁矿(α-Fe2O3)结构,且纯的Fe2O3具有较好的结晶度(JCPDS 33-0644)[23]。此外,具有不同RuO2含量的RuO2-Fe2O3催化剂表现出与纯的Fe2O3相似的衍射峰。在XRD谱图中既没有观察到金属钌物种的峰也没有观察到氧化钌物种的出现。由此可见,低含量的氧化钌物种高度分散于催化剂表面以至于不能被XRD检测到,或者是氧化钌物种表现出无定型的结构[24]。
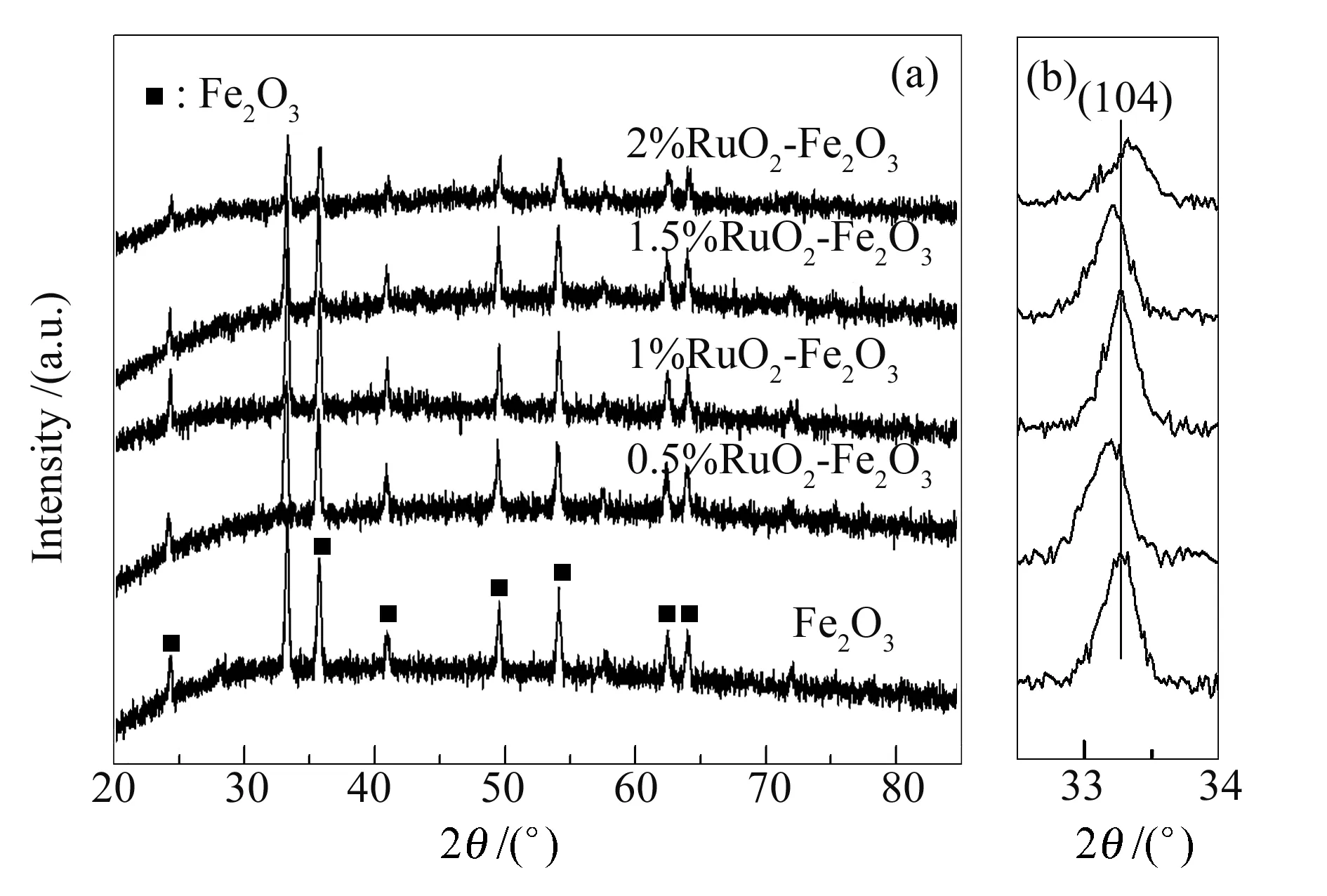
图 2 纯的Fe2O3和RuO2-Fe2O3催化剂的XRD谱图
为进一步研究氧化钌掺杂对Fe2O3结构的影响,对(104)晶面的衍射峰进行了放大处理,结果见图2(b)。随着氧化钌的掺杂,当RuO2含量为2%时,位于33.3°处的Fe2O3的衍射峰发生了轻微的高偏(0.3°)。可能是由于具有较小金属离子半径的Ru物种(Ru4+:0.062 nm))部分成功掺杂进入具有较大金属离子半径的Fe2O3晶格结构中(Fe3+:0.065 nm),导致了Fe2O3晶格参数的改变[25]。但是,低含量(<2%)的RuO2基催化剂并没有发生这一高偏现象,可能是由于钌物种以氧化钌的形式分布在Fe2O3表面或孔道内。
2.3 BET分析
图3为Fe2O3和RuO2-Fe2O3催化剂的N2吸脱附曲线图(图3(a))和相应的孔径分布图(图3(b))。

图 3 不同催化剂的N2吸脱附曲线(a)和孔径分布图(b)
由图3(a)可知,所有样品的等温线都属于类型Ⅳ,滞回环为H3型,根据IUPAC分类标准可知,所有样品均为介孔材料[26]。孔径分布曲线结果表明,纯的Fe2O3样品的孔径集中分布在48 nm,少量氧化钌(<2%)的掺杂使催化剂的孔径分布变化不大。结合表1中催化剂的孔容和比表面积结果可以看出,少量氧化钌的加入对催化剂孔结构影响不是很明显,这可能与氧化钌物种的高度分散有关[18]。而当氧化钌含量增加到2%时,催化剂孔径分布明显减小(38 nm),孔容轻微增加,这也是其比表面积增大的原因。此外,造成孔径减小的原因可能是氧化钌除了部分进入到Fe2O3晶格中,还有一部分堆积在催化剂表面形成一些稍小的孔;另一方面,可能是在焙烧过程中形成更多比纯的Fe2O3稍小的孔。由此可见,RuO2含量对样品的孔径分布影响较大。
表1为Fe2O3和RuO2-Fe2O3催化剂的比表面积和孔容信息。由表1可知,纯的Fe2O3的比表面积为14.3 m2/g。此外,除了1%RuO2-Fe2O3催化剂外,其他催化剂的比表面积与纯的Fe2O3相比都有所增大,当RuO2的含量为2%时,比表面积最大,结合活性测试结果可知,大的比表面积有利于活性位点的分布,从而提高了催化剂的催化活性。

表 1 不同催化剂的孔结构参数
2.4 H2程序升温还原(H2-TPR)
根据H2-TPR谱图中催化剂还原峰出现的温度以及还原峰的强度可以测定催化剂的氧化还原能力,图4为Fe2O3和RuO2-Fe2O3催化剂的H2-TPR谱图。由图4可知,纯的Fe2O3样品出现了两个还原峰:其中,出现在380 ℃左右的低温还原峰归属于Fe2O3到Fe3O4的还原;起始位置高于600 ℃的高温还原峰则归属于Fe3O4到 FeO 和Fe0的进一步还原[27]。
此外,与纯的Fe2O3相比,RuO2-Fe2O3催化剂中属于Fe2O3和Fe3O4的还原峰,位置明显向低温偏移,说明RuO2的加入促进了Fe物种的还原,同时也说明发生了从Ru原子向氧化铁的氢溢流现象[28]。其中,钌物种对铁物种还原能力的促进作用表明RuO2与Fe2O3之间存在一种协同效应,这种协同效应提高了催化剂的还原能力。结合活性测试结果可以看出,还原能力的提高促进了催化剂的氧化反应。
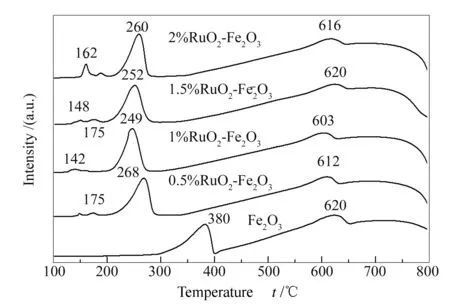
图 4 不同催化剂的H2-TPR谱图
由图4还可知,随着氧化钌的加入,出现了一个温度更低的还原峰,并且随着氧化钌含量的增加,该还原峰的强度逐渐增强,氧化钌含量最高的样品强度变化最明显。由此可以推断该还原峰为氧化钌物种的还原。
2.5 NH3程序升温脱附(NH3-TPD)
Fe2O3和RuO2-Fe2O3催化剂的NH3-TPD实验结果见图5。

图 5 不同催化剂的NH3-TPD谱图
根据先前文献报道,低于200 ℃的NH3脱附峰属于弱吸附的NH3物种,尤其是脱附温度低于100 ℃时,可能是由于物理吸附或弱吸附的NH3物种造成的[29,30];而脱附温度高于250 ℃的脱附峰一般属于中强酸性位点上吸附氨物种的脱附[30]。从图5纯的Fe2O3样品中可以看出,Fe2O3主要存在两个脱附峰,一个归属于物理吸附或弱吸附NH3物种的脱附峰,另一个归属于中强酸性位点上吸附氨物种的脱附。随着RuO2的加入,催化剂表面的酸性环境发生明显变化。当RuO2含量大于1%时,中强酸性位点上脱附的氨物种明显增多,表现在其脱附峰峰强度明显增强。除此之外,所有RuO2-Fe2O3催化剂的弱酸性位点相比于纯的Fe2O3也有所增加。众所周知,酸性位点的增加有利于NH3的吸附,使更多的NH3能够被吸附并参与被活性金属活化的反应。结合活性测试结果可知(图1),RuO2作为主要的活性位点,它的加入不仅促进了NH3的吸附,也提高了NH3与表面活跃氧发生氧化反应的能力,这一结论将在接下来的原位漫反射红外光谱研究中得到进一步证明。
2.6 NH3吸附的原位漫反射红外光谱研究
原位漫反射红外光谱实验进一步说明了催化剂表面酸性位点的种类以及随时间的变化情况。图6为250 ℃时NH3在Fe2O3和RuO2-Fe2O3催化剂表面随吸附时间变化的DRIFTS实验结果。

图 6 250 ℃时NH3在不同催化剂表面吸附变化的原位漫反射红外光谱谱图

对比图6(a),图6(b)-6(e)实验结果主要研究RuO2-Fe2O3催化剂中不同RuO2含量对Fe2O3表面酸性位点的影响以及NH3吸附的变化情况。所有RuO2-Fe2O3样品均在1822 cm-1处观察到一个新的吸附峰[7,34]。根据文献结果可知,该吸附峰归属于亚硝酰基(-HNO)物种。此外,当RuO2含量为0.5%和2%时,归属于Lewis酸性位点峰的位置发生了偏移,分别偏移至1585 和1588 cm-1[35]。与Fe2O3相比,RuO2-Fe2O3催化剂的另一个差异主要体现在酸强度的变化上。随着RuO2含量的增加,RuO2-Fe2O3催化剂上Lewis酸性位点的峰强度逐渐减弱,亚硝酰基(-HNO)物种的峰强度增强。其中,RuO2含量为1.5%和2%时变化最明显。然而,RuO2含量的进一步增加导致Fe2O3表面的Brønsted酸性位点消失。
以上研究结果表明,RuO2的掺杂提高了催化剂表面晶格氧的活化。一方面体现在:结合NH3-TPD结果可知,RuO2的加入使催化剂表面的酸性位点有所增加,但是在DRIFTS图中反而观察到吸附氨物种的L酸性位点和B酸性位点出现减少的趋势;另一方面,-HNO物种的出现也说明了吸附的NH3物种被活化并发生了氧化反应。总的来说,RuO2-Fe2O3催化剂上吸附在酸性位点上的NH3快速发生脱氢反应生成酰胺(-NH2)物种,-NH2进一步脱氢形成亚酰胺(-NH)物种,导致红外光谱监测到的吸附氨物种减少[36]。之后,-NH与催化剂上被RuO2活化的活跃原子氧发生快速氧化反应生成-HNO物种。主要的脱氢反应和氧化反应如下:
NH3→ NH2+ H
(7)
NH2→ NH + H
(8)
NH + O → HNO
(9)
整个过程发生的速率很快,以致某些步骤并不能很好的被红外光谱实验监测到。同时,结合活性测试结果证实了RuO2-Fe2O3催化剂表面存在更多的活性位点,导致吸附的NH3物种与催化剂表面的活跃氧物种发生氧化反应,这也是RuO2-Fe2O3催化剂活性好的原因。此外,根据先前文献报道[7,36],-HNO物种一般是原位选择性催化还原反应(i-SCR)的中间产物,即-HNO进一步被氧化为NO物种,NO再与酰胺(-NH2)反应生成N2。而i-SCR反应机理一般认为是NH3-SCO反应中较为常见的反应途径。
3 结 论
与Fe2O3相比,RuO2-Fe2O3催化剂表现出较好的低温催化活性,并且RuO2含量对催化剂的活性影响明显,1.5%RuO2-Fe2O3和2%RuO2-Fe2O3均在225 ℃实现了NH3的完全转化。H2-TPR结果表明,RuO2与Fe2O3之间的协同效应有效地提高了催化剂的催化活性。此外,NH3-TPD结果表明RuO2的掺杂提高了催化剂表面的中强酸性位点,且RuO2含量越高酸性变化越明显。DRIFTS研究进一步显示催化剂表面主要存在Lewis酸性位点,在活跃的RuO2-Fe2O3表面发生了脱氢和氧化反应:吸附在Lewis酸性位点上的NH3物种发生脱氢反应形成-NH中间物种,-NH物种与催化剂表面上被钌物种活化的活跃氧物种发生快速的氧化反应,生成-HNO物种。
