合成条件对磷钨酸负载的金属有机框架催化剂氧化脱硫性能的影响
2019-03-06,,,*,,
, ,,*, ,
(1.东北石油大学 化学化工学院, 黑龙江 大庆 163318;2.东北石油大学 石油与天然气化工省重点实验室, 黑龙江 大庆 163318)
油品中的含硫化合物,如硫化氢、硫醇、硫醚及噻吩类硫化物,其燃烧形成的SO2、SO3等酸性气体对环境和人们的健康造成严重的破坏[1,2]。因此,深度脱硫技术已成为全世界关注的热点之一。目前,加氢脱硫技术已经广泛应用于工业生产中,但加氢脱硫工艺对无机硫和部分有机硫化合物有较好的脱除效果,但对于稠环噻吩类含硫化合物及其衍生物的脱除比较困难,需要高温、高压,且对设备要求相应较高,难以达到低成本深度脱硫的目的。相比之下,氧化脱硫技术条件温和、脱硫率高且成本较低,适合深度脱硫[3]。因此,氧化脱硫技术以其显著的优点引起了研究者的极大关注。
杂多酸[4,5]作为一类具有高氧化还原性、高催化活性的固体杂多酸催化剂,在氧化脱硫领域具有极大的研究价值。2009年,Li等[6]分别以1-丁基-3-甲基咪唑四氟硼酸盐、六氟硼酸盐和1-辛基-3-甲基咪唑四氟硼酸盐、六氟硼酸盐为催化剂,以过氧化氢为氧化剂,探究了加入磷钨酸对二苯并噻吩(DBT)萃取氧化脱硫性能的影响。结果表明,以单一离子液体为催化剂时,对DBT转化率不足30%,而加入磷钨酸构成的双催化剂体系时,DBT转化率可上升至98.2%,表明磷钨酸对氧化脱硫具有很好的催化作用。然而,杂多酸存在比表面积低、分散性差、易溶于反应体系,使其的分离和回收较难等缺点[7]。因此,解决杂多酸分散性差及回收、分离困难的问题是该领域研究的是关键问题。
近年来,金属有机框架(MOFs)因其高孔隙率、结构可调、比表面积大等优势被广泛应用于催化反应中[8]。较大比表面积的MOFs作为杂多酸的载体可以弥补杂多酸团聚造成的不足,同时有效解决催化剂的回收困难等问题。Ding等[9]制备了POM@MOF,并以O2为氧化剂,催化氧化DBT,在最佳反应条件下,对DBT的脱硫率可达97%,重复利用三次后活性仅轻微降低。
本实验通过一步合成法合成了磷钨酸负载的金属有机框架HPW@MIL-101(Cr)复合型催化剂。以过氧化氢为氧化剂,探究了合成时间、合成温度、酸碱度对HPW@MIL-101(Cr)复合型催化剂结构以及氧化脱硫活性的影响。
1 实验部分
1.1 实验药品
N,N-二甲基甲酰胺、无水乙醇(辽宁泉瑞试剂有限公司);九水合硝酸铬((Cr(NO3)3·9H2O)、磷钨酸(HPW)(上海麦恪林生化科技有限公司);对苯二甲酸(H2BDC)(上海阿拉丁试剂有限公司);过氧化氢 (H2O2, 30%)、正辛烷(天津市大茂化学试剂厂);苯并噻吩(BT)(百灵威试剂 USA)。
1.2 催化剂的制备
将 Cr(NO3)3·9H2O、H2BDC和Z(中性的水)按1∶1∶280的物质的量比混合,然后加入3.5 g HPW至250 mL烧杯中,磁力搅拌。转移至不锈钢内衬聚四氟乙烯反应釜内,使其充分扩散至水溶液中。将准备好的不锈钢内衬聚四氟乙烯反应放入温度为220 ℃的真空干燥箱中,分别加热2、4、8、12 h后,冷却至室温。取出固液混合物,抽滤,分别用N,N-二甲基甲酰胺和乙醇清洗,150 ℃干燥,得到的不同合成时间的绿色固体催化剂用HPW(3.5)@MIL-101(Cr)-x-y-z表示,其中,x表示合成时间(h),y为合成温度(℃),z为所用溶液的酸碱度(酸性用s表示,中性zh表示,碱性用j表示)。
合成时间12 h及其他制备条件不变(同上)的前提下,设置不锈钢内衬聚四氟乙烯反应釜内的合成温度分别为100、140、180和220 ℃,获得不同合成温度的催化剂,记为HPW(3.5)@MIL-101(Cr)-12-y-zh。
在合成时间12 h和合成温度220 ℃及其他制备条件不变(同上)的前提下,分别用质量分数为0.5%的酸性氢氟酸(HF)和碱性四甲基氢氧化胺(TMAOH)水溶液替代,获得不同酸碱度的催化剂,记为HPW(3.5)@MIL-101(Cr)-12-220-z。
在合成时间12 h、合成温度220 ℃、所用溶液为水溶液及其他制备条件不变(同上)的前提下,改变HPW负载量(分别为0.5、2.0、3.5和5.0 g),获得不同HPW负载量的催化剂,记为HPW(a)@MIL-101(Cr)-12-220-h,其中,a表示HPW负载量(g)。
1.3 催化剂的表征
波长400-4000 cm-1的Tensor 27傅里叶红外光谱仪;通过TRISTAR II 3020微粒吸附设备及BET法对催化剂的物理化学性质进行分析;在CuKα辐射扫描1°-10°并以1(°)/min为扫描速率的条件下,通过D/max-2200PC-X-ray仪器测得X射线衍射数据;模拟油中硫化物的含量通过Clarus 680气相色谱测定。
1.4 催化剂的性能评价
1.4.1模拟油的配置
称取一定量的苯并噻吩(BT),噻吩(TP),二苯并噻吩(DBT)和4,6-二甲基苯并噻吩(4,6-DMDBT)分别溶于正辛烷后,在四个250 mL 容量瓶中分别定容,得到BT、TP、DBT和4,6-DMDBT的含量为500 mg/L 的模拟油。
1.4.2脱硫率的测定方法
实验中通过Clarus 680 GC-FPD珀金埃尔默气相色谱仪对硫含量的进行测定,FPD检测器进行检测。具体硫含量测定的分析条件如下:FPD检测器温度为 250 ℃,进样口温度为250 ℃,柱箱程序升温如下:初始温度为60 ℃,保留1 min,以10 ℃min升温至250 ℃,保留2 min,随后降低至室温。
1.4.3脱硫率计算方法
η表示脱硫率,C0表示为苯并噻吩模拟油的初始浓度,Ct表示为氧化脱硫反应进行至某一时刻的苯并噻吩模拟油中的硫浓度。脱硫率计算方法如公式(1):

(1)
2 结果与讨论
2.1 催化剂的表征
2.1.1XRD分析
图1为不同合成时间制备的HPW(3.5)@MIL-101(Cr)-x-220-zh催化剂的XRD谱图。由图1可知,各催化剂在2.80°、3.30°、3.45°、4.00°处均出现了MIL-101(Cr)的尖锐的衍射峰,表明催化剂晶型为MIL-101(Cr)相[10]。这一结果表明,不同合成时间制备的催化剂中,MIL-101(Cr)的晶体结构良好。各催化剂的特征峰强度由强到弱的顺序为HPW(3.5)@MIL-101(Cr)-12-220-zh > HPW(3.5)@MIL-101(Cr)-8-220-zh > HPW(3.5)@MIL-101(Cr)-4-220-zh > HPW(3.5)@MIL-101(Cr)-2-220-zh,表明随着合成时间的延长,催化剂中MIL-101(Cr)的孔道有序度逐渐提高。

图 1 不同合成时间制备的催化剂的XRD谱图
图2为合成时间12 h等其他条件不变的前提下,不同合成温度制备的HPW(3.5)@MIL-101(Cr)-12-y-zh催化剂的XRD谱图。由图2可知,合成温度为220及180 ℃的HPW(3.5)@MIL-101(Cr)-12-220-zh和HPW(3.5)@MIL-101(Cr)-12-180-zh催化剂的谱图中,在2.80°、3.30°、3.45°、4.00°处出现了MIL-101(Cr)的衍射峰,并且随着合成温度升高,其孔道有序度逐渐提高。合成温度为140、100 ℃的HPW(3.5)@MIL-101(Cr)-12-140-zh和HPW(3.5)@ MIL-101(Cr)-12-100-zh催化剂的谱图中,未见金属有机框架MIL-101(Cr)的特征峰,表明催化剂合成温度≤140 ℃时,不能形成MIL-101(Cr)晶体结构。

图 2 不同合成温度制备的催化剂XRD谱图
图3为合成时间12 h、合成温度220 ℃等其他条件不变的前提下,不同酸碱度的HPW(3.5)@MIL-101(Cr)-12-220-z催化剂的XRD谱图。在酸性、中性、碱性条件下合成的HPW(3.5)@MIL-101(Cr)-12-220-s、HPW(3.5)@MIL-101(Cr)-12-220-zh和HPW(3.5)@MIL-101(Cr)-12-220-j催化剂在2.80°、3.30°、3.45°、4.00°处均出现了MIL-101(Cr)的衍射峰,表明酸性、中性、碱性环境下合成的催化剂均能得到MIL-101(Cr)晶体结构。对比各催化剂的特征峰强度,可以明显看到在酸性条件下制备的HPW(3.5)@MIL-101(Cr)-12-220-s催化剂的特征峰强度要弱于中性及碱性条件下制备的HPW(3.5)@MIL-101(Cr)-12-220-zh、HPW(3.5)@MIL-101(Cr)-12-220-j催化剂。说明酸性条件下HPW@MIL-101(Cr)的孔道有序度降低。

图 3 不同酸碱度催化剂的XRD谱图
2.1.2FT-IR分析
图4为不同合成时间制备的HPW(3.5)@MIL-101(Cr)-x-220-zh催化剂的FT-IR谱图。
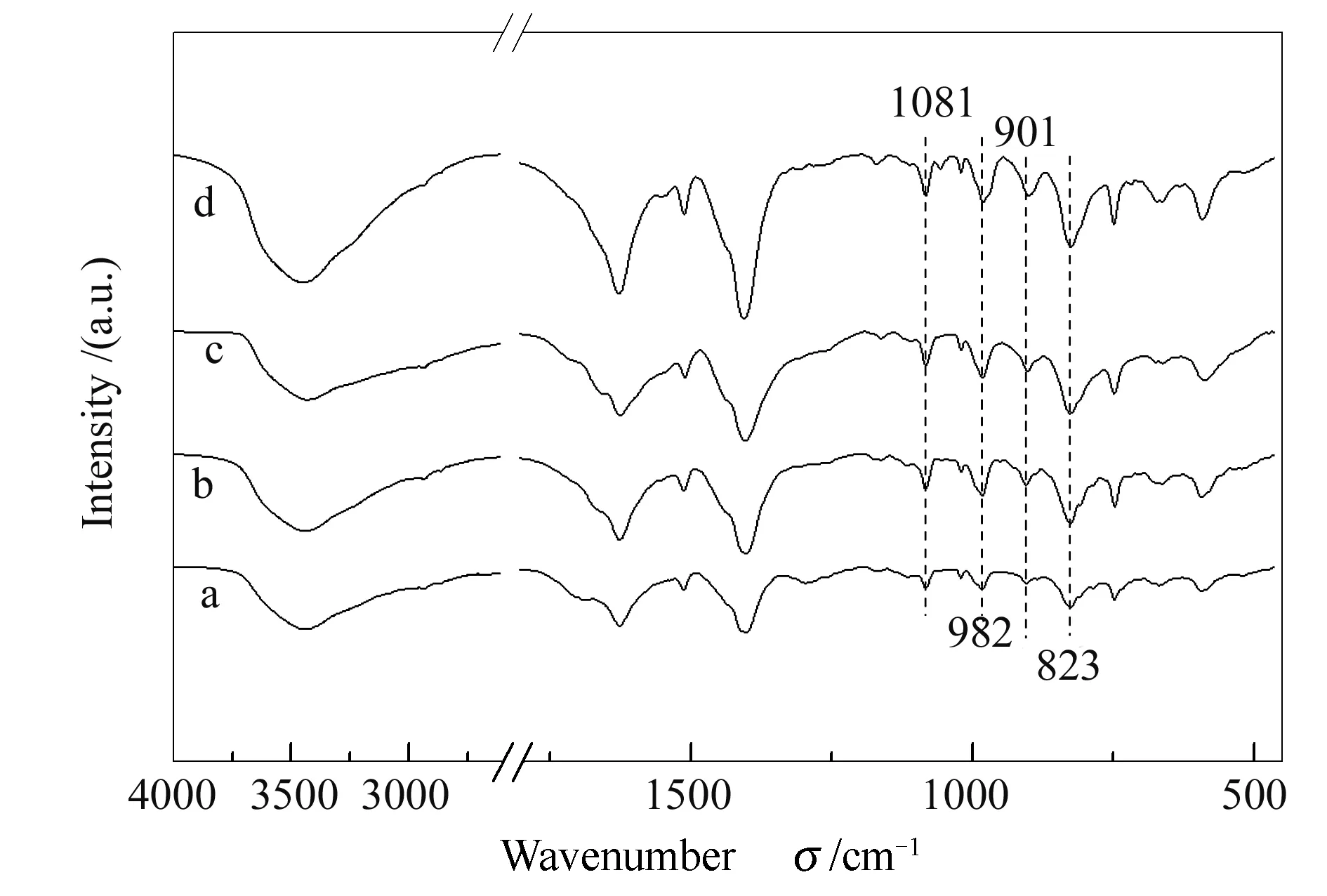
图 4 不同合成时间制备的催化剂的FT-IR谱图
各催化剂在1200-1800 cm-1处出现了四指型MIL-101(Cr)的伸缩振动峰[11],表明不同合成时间制备的HPW(3.5)@MIL-101(Cr)-x-220-zh催化剂中金属有机框架的结构完整;在1081、982、901和823 cm-1处出现的特征峰,分别对应着具有Keggin结构的P-Oa、W=Od、W-Ob-W和W-Oc-W的伸缩振动峰[12],表明各催化剂中均保留有磷钨酸的Keggin结构特征峰,证实了在给定的不同合成时间下,均能将磷钨酸成功负载到金属有机框架,从而得到HPW@MIL-101(Cr)催化剂。
图5为合成时间12 h等其他条件不变的前提下,不同合成温度制备的HPW(3.5)@MIL-101(Cr)-12-y-zh催化剂的FT-IR谱图。合成温度为220、180 ℃的HPW(3.5)@MIL-101(Cr)-12-220-zh和HPW(3.5)@MIL-101(Cr)-12-180-zh催化剂在1200-1800 cm-1处出现四指型MIL-101(Cr)的伸缩振动峰;在1081、982、901和823 cm-1(P-Oa、W=Od、W-Ob-W和W-Oc-W)处出现独特的Keggin结构特征峰,表明催化剂中保留有磷钨酸的Keggin结构特征峰,这说明温度高于180 ℃时,磷钨酸可成功负载至金属有机框架,从而得到HPW@MIL-101(Cr)催化剂(XRD分析,图2)。合成温度为140、100 ℃的HPW(3.5)@MIL-101(Cr)-12-140-zh和HPW(3.5)@MIL-101(Cr)-12-100-zh催化剂的谱图中,并未出现MIL-101(Cr)与Keggin结构特征峰,说明在合成温度为140 ℃及以下不能获得具有MIL-101(Cr)晶体结构的HPW@MIL-101(Cr)催化剂,此结果与XRD分析结果一致。

图 5 不同合成温度制备的催化剂的FT-IR谱图
图6为合成时间12 h、合成温度220 ℃等其他条件不变的前提下,不同酸碱度的HPW(3.5)@MIL-101(Cr)-12-220-z催化剂的FT-IR谱图。酸性、中性、碱性条件下制备的HPW(3.5)@MIL-101(Cr)-12-220-s、HPW(3.5)@MIL-101(Cr)-12-220-zh、HPW(3.5)@MIL-101(Cr)-12-220-j催化剂在1200-1800 cm-1处均出现四指型MIL-101(Cr)的伸缩振动峰。同时,在1081、982、901和823 cm-1(P-Oa、W=Od、W-Ob-W和W-Oc-W)处均出现独特的Keggin结构特征峰,表明催化剂在酸性、中性、碱性合成条件下均可形成结构良好的MIL-101(Cr)晶体,且保留Keggin结构特征峰。

图 6 不同酸碱度催化剂的FT-IR谱图
2.1.3N2吸附数据分析
表1为不同合成时间、温度、酸碱度和HPW负载量条件下制备获得的HPW(a)@MIL-101(Cr)-x-y-z催化剂的N2吸附数据。由表1可知,HPW的比表面积、孔隙容积、平均孔径分别为13 m2/g、10.46 cm3/g、17.9 nm。MIL-101(Cr)的比表面积、孔隙容积、平均孔径分别为2465 m2/g、0.93 cm3/g、3.1 nm。HPW负载量分别为0.5、2.0、3.5和5.0 g的HPW(a)@MIL-101(Cr)-12-220-z催化剂的比表面积分别为1475、1213、1054和916 m2/g。与MIL-101(Cr)相比,催化剂负载HPW后,比表面积均显著减小,同时伴随有孔容和孔径的减小。这表明HPW进入到金属有机框架的孔道内,被封装至MIL-101(Cr)空腔内部。随着HPW负载量的增加,催化剂比表面积减小,进一步表明随着HPW负载量的增加,进入MIL-101(Cr)孔道的HPW越多,从而导致比表面积的逐渐减小。此外,与未负载的HPW相比,催化剂HPW@MIL-101(Cr)的比表面积明显增大,金属有机框架巨大的比表面积确保了HPW在其表面高度分散,从而提供更多的活性位点[13]。
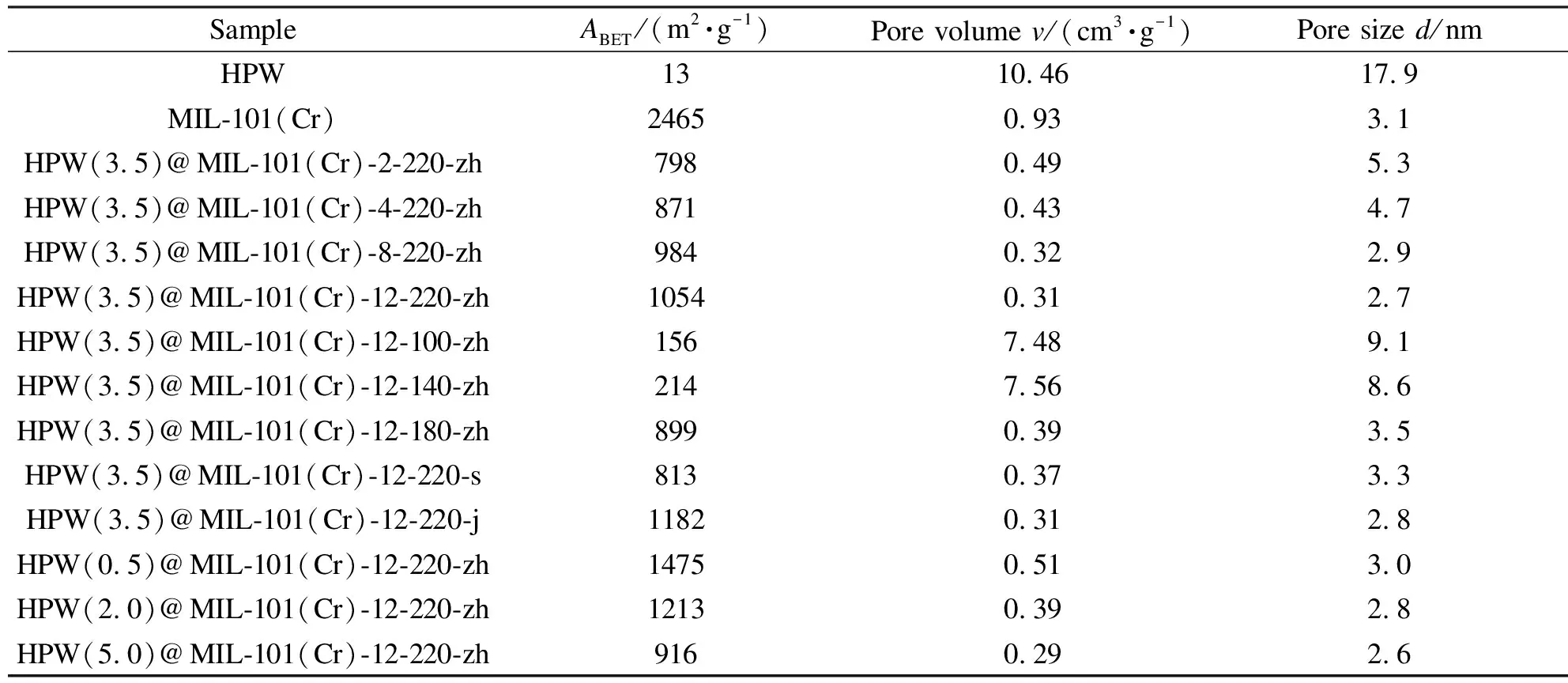
表 1 催化剂的N2吸附数据
随着合成时间的延长,HPW(3.5)@MIL-101(Cr)-x-220-zh的比表面积逐渐增大,孔隙容积及平均孔径逐渐减小,这主要是由于合成时间过短时,金属有机框架的孔道有序度低(XRD分析,图1),其孔道形成不完整所致。
合成温度为100、140 ℃制备的HPW(3.5)@MIL-101(Cr)-12-100-zh和HPW(3.5)@MIL-101(Cr)-12-140-zh催化剂的比表面积分别为156 m2/g和214 m2/g,与MIL-101(Cr)相比比表面积减小显著,这主要是由于低的合成温度(100、140 ℃)下获得的催化剂,没有形成大比表面积的MIL-101(Cr)晶体结构(XRD分析,图2;FT-IR分析,图5)所致。合成温度为180和220 ℃制备的HPW(3.5)@MIL-101(Cr)-12-180-zh和HPW(3.5)@MIL-101(Cr)-12-220-zh催化剂的比表面积分别为899和1054 m2/g,且随着合成温度的提高比表面积增大。这是由于随着合成温度的升高,金属有机框架孔道有序度提高(XRD分析,图2),催化剂孔道形成更加完整,从而增大了催化剂的比表面积。
对比不同酸碱合成条件下制备的HPW(3.5)@MIL-101(Cr)-12-220-z催化剂,碱性和中性环境下制备的HPW(3.5)@MIL-101(Cr)-12-220-j及HPW(3.5)@MIL-101(Cr)-12-220-zh催化剂的比表面积分别为1182和1054 m2/g,其比表面积均大于1000 m2/g。而酸性环境下制备的HPW(3.5)@MIL-101(Cr)-12-220-s催化剂的比表面积只有813 m2/g,与HPW(3.5)@MIL-101(Cr)-12-220-j及HPW(3.5)@MIL-101(Cr)-12-220-zh催化剂相比,比表面积显著减小。这主要是由于酸性合成环境下,催化剂中MIL-101(Cr)的孔道有序度降低(XRD分析,图3),导致的比表面积的减小。
2.2 催化剂的活性评价
2.2.1合成时间的影响
图7为合成时间对HPW(3.5)@MIL-101(Cr)-x-220-zh催化剂BT催化氧化脱硫性能的影响。

图 7 合成时间对HPW(3.5)@MIL-101(Cr)-x-220-zh催化剂BT催化氧化脱硫性能的影响
反应条件为BT模拟油20 mL、催化剂用量0.24 g、氧硫比8∶1、反应温度50 ℃、反应时间120 min(没有特殊说明,后面的讨论中反应条件不变)。由图7可知,HPW(3.5)@MIL-101(Cr)-2-220-zh、HPW(3.5)@MIL-101(Cr)-4-220-zh、HPW(3.5)@MIL-101(Cr)-8-220-zh和HPW(3.5)@MIL-101(Cr)-12-220-zh催化剂对BT脱硫率分别为62%、87%、93%、99%,表明随着催化剂合成时间的延长,其脱硫效果逐渐提高,合成时间为12 h制备的HPW(3.5)@MIL-101(Cr)-12-220-zh催化剂表现出最佳的催化活性。这主要是由于HPW(3.5)@MIL-101(Cr)-12-220-zh催化剂的比表面积较大(BET分析,表1),HPW在金属有机框架上的分散度高,从而提高了催化剂催化性能。
2.2.2合成温度的影响
图8为合成温度对HPW(3.5)@MIL-101(Cr)-12-y-zh催化剂BT催化氧化脱硫性能的影响。由图8可知,合成温度为100和140 ℃制备的HPW(3.5)@MIL-101(Cr)-12-100-zh、HPW(3.5)@MIL-101(Cr)-12-140-zh催化剂对BT脱硫率分别为20%、23%,催化活性较低。这主要是由于合成温度过低,催化剂未形成MIL-101(Cr)晶体结构,催化剂比表面积小(BET分析,表1),HPW的分散不好,从而影响了催化剂的催化性能。合成温度为180和220 ℃制备的HPW(3.5)@MIL-101(Cr)-12-180-zh和HPW(3.5)@MIL-101(Cr)-12-220-zh催化剂对BT脱硫率分别为78%和99%,催化氧化脱硫效果明显优于HPW(3.5)@MIL-101(Cr)-12-100-zh、HPW(3.5)@MIL-101(Cr)-12-140-zh。这主要是良好的MIL-101(Cr)晶体结构极大地提高了催化剂的比表面积(BET分析,表1),表明大比表面积金属有机框架结构,对提高HPW催化性能具有至关重要的作用。

图 8 合成温度对HPW(3.5)@MIL-101(Cr)-12-y-zh催化剂BT催化氧化脱硫性能的影响
2.2.3酸碱度的影响
表2为酸碱度对HPW(3.5)@MIL-101(Cr)-12-220-z催化剂BT催化氧化脱硫性能的影响。由表2可知,HPW(3.5)@MIL-101(Cr)-12-220-s、HPW(3.5)@MIL-101(Cr)-12-220-zh和HPW(3.5)@MIL-101(Cr)-12-220-j催化剂对BT脱硫率分别为68%、99%、85%,在中性条件下合成的HPW@MIL-101(Cr)催化剂具有最佳脱硫性能。HPW(3.5)@MIL-101(Cr)-12-220-s催化性能最低,这是由于酸性条件下合成的催化剂的孔道有序度低、比表面积小(BET分析,表1)的缘故;而HPW(3.5)@MIL-101(Cr)-12-220-j催化剂催化氧化脱硫效果不佳,推测可能是碱性环境下合成HPW@MIL-101(Cr)催化剂对具有酸性性质的活性组分HPW的有一定的影响,从而导致催化活性下降。

表 2 HPW(3.5)@MIL-101(Cr)-12-220-z催化剂BT催化氧化脱硫性能的影响
2.2.4HPW负载量的影响
图9为HPW负载量对HPW(a)@MIL-101(Cr)-12-220-zh催化剂BT催化氧化脱硫性能的影响。HPW负载量分别为0.5、2.0、3.5和5.0 g的HPW(a)@MIL-101(Cr)-12-220-z催化剂对BT脱硫率分别为65%、82%、99%、87%。表明随着HPW负载量的增加,HPW(a)@MIL-101(Cr)-12-220-zh催化剂的催化性能呈现先升高后降低的趋势。当HPW负载量为3.5 g时,HPW(3.5)@MIL-101(Cr)-12-220-zh催化剂具有最佳催化活性。这可能是由于负载量的增加,催化剂拥有更多的活性位点,有助于提高催化活性。然而,过量的HPW负载量会导致催化剂表面积减小(见表1),堵塞催化剂的孔道,反而不利于提高催化剂的活性。

图 9 HPW负载量对HPW(a)@MIL-101(Cr)-12-220-zh催化剂BT催化氧化脱硫性能的影响
2.2.5催化剂脱硫性能的比较
图10为BT模拟油20 mL、催化剂用量0.24 g、氧硫比8∶1、反应温度50 ℃反应条件下,HPW(3.5)@MIL-101(Cr)-12-220-zh与HPW催化剂脱硫性能的比较。氧化脱硫时间为30 min时,HPW催化剂对BT脱硫率仅为4%,随着反应时间的延长,脱硫率提高,在反应时间120 min时,对BT的脱硫率达到28%。而HPW(3.5)@MIL-101(Cr)-12-220-zh催化剂,反应时间为30 min时,对BT脱硫率已达到68%,与HPW为催化剂时相比,脱硫率提高了16.0倍;继续延长反应时间至120 min时,HPW(3.5)@MIL-101(Cr)-12-220-zh脱硫率高达99%,与HPW为催化剂时相比,脱硫率提高了2.4倍。表明HPW负载于大比表面积的MIL-101(Cr)后,能够明显增大催化剂的比表面积,改善HPW分散性,从而提供更多的活性位点。此外,金属有机框架的孔道结构有助于反应物和产物的扩散,是HPW(3.5)@MIL-101(Cr)-12-220-zh拥有高的脱硫活性的另一原因(BET分析,表1)。

图 10 HPW(3.5)@MIL-101(Cr)-12-220-zh与HPW脱硫性能比较
2.2.6不同模拟油对脱硫率的影响
表3为HPW(3.5)@MIL-101(Cr)-x-220-zh催化剂对不同噻吩类硫化物催化氧化脱硫性能的影响。反应条件为模拟油20 mL、催化剂用量0.24 g、氧硫比8∶1、反应温度50 ℃、反应时间120 min。由表3可知,催化剂对TP模拟油的脱硫率只有58%,脱硫率最低,这主要是由于TP的电子云密度低,从而导致对其脱硫效果最差[14]。催化剂对BT、DBT、4,6-DMDBT模拟油的脱硫率分别为99%、100%、99%。表明HPW@MIL-101(Cr)对这些噻吩类硫化物均具有较好的脱硫效果。

表 3 其他油品的氧化脱硫性能
3 结 论
随着合成时间的延长、合成温度升高,HPW@MIL-101(Cr)催化剂孔道有序度提高;合成温度≤140 ℃时,不能形成MIL-101(Cr)晶体结构;酸性合成条件合成的HPW@MIL-101(Cr)催化剂的孔道有序度均比中性和碱性条件合成的催化剂低。
随着合成时间的延长、合成温度升高,HPW@MIL-101(Cr)催化剂的脱硫活性提高;中性条件下合成的HPW@MIL-101(Cr)催化剂的催化性能优于酸性和碱性条件下合成的催化剂。
随着HPW负载量的增加,HPW@MIL-101(Cr)催化剂对BT脱硫效果呈现先升高后降低的趋势。
在12 h、220 ℃、中性条件下合成的负载量为3.5 g的HPW@MIL-101(Cr)催化剂具有最佳的催化氧化脱硫活性。在模拟油20 mL、催化剂用量0.24 g、氧硫比8∶1、反应温度50 ℃、反应时间为120 min时,对BT脱硫率高达99%。
在相同条件下,氧化脱硫时间为30 min时,本文制备的HPW@MIL-101(Cr)催化剂对BT脱硫率是纯HPW催化剂的17.0倍;氧化脱硫时间为120 min时,HPW@MIL-101(Cr)催化剂对BT脱硫率是纯HPW催化剂的3.4倍,表明将HPW负载于金属有机框架MIL-101(Cr),能够显著提高HPW催化剂的脱硫活性。
HPW@MIL-101(Cr) 催化剂对TP脱硫率只有58%,但对DBT和4,6-DMDBT均具有良好的脱硫活性,脱硫率分别为100%和99%。
