主动脉夹层中层P53、MDM2及TRIM25的表达变化和VSMC表型转化
2019-03-05王志维李博文昌金星王嘉慧
任 伟 王志维 李博文 昌金星 王嘉慧
主动脉夹层(aortic dissection,AD)是心血管外科的常见的灾难性疾病之一,其发生率呈逐年上升趋势,目前认为主动脉中层平滑肌的退行性病变、炎症、力学冲击三者参与夹层的发生,平滑肌收缩对血流力学冲击的拮抗是血管的稳态之一,一旦损伤容易诱发夹层。血管平滑肌细胞(vascular smooth muscle cells,VSMC)的表型转化是主动脉中层退行性变的主要特征,但是其转化的具体机制仍未阐明[1~3]。三基序蛋白25(tripartite-motif protein25, TRIM25)能够通过泛素化参与多种细胞功能的调节,包括细胞周期调控和转录,主要在子宫、胚胎及主动脉组织中表达[4,5]。本研究通过对正常主动脉组织和夹层主动脉中层中TRIM25、P53、MDM2表达量变化和VSMC表型变化的研究,探讨其在AD形成过程的作用及可能机制。
材料与方法
1.一般资料:选取2015年1月~2016年1月在武汉大学人民医院行全弓替换手术患者12例作为AD组。纳入标准:主动脉CTA明确诊断为A型主动脉夹层,发病1周以内;排除标准:马方综合征和特纳综合征等结缔组织疾病,合并二叶式主动脉瓣畸形或胸部损伤,再次心脏手术者和CTA或术中发现严重的主动脉壁粥样硬化。正常对照组的患者来自于心脏死亡捐献者,无主动脉疾病史,其他排除标准同上。术前资料总结发现两组患者年龄性别和体重无明显差异,AD组合并高血压者明显高于正常组,详见表1。术前签署知情同意书,并获得武汉大学人民医院伦理委员会的批准。
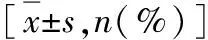
表1 AD组和正常对照组的基本特征
主要试剂骨桥蛋白(OPN)和(平滑肌肌动蛋白(α-SMA)兔单克隆抗体购于北京博奥森公司,P53和p-P53(Ser15) 鼠抗人单克隆抗体购于美国Santa Cruz公司,MDM2兔抗人单克隆抗体购于英国Abcam公司,TRIM25兔抗人单克隆抗体购于武汉三鹰公司,抗鼠/兔二抗购于美国Licor公司。
2.免疫组化染色:获取升主动脉组织后去除主动脉外膜和内膜,保留中层组织,固定后石蜡包埋,4μm厚切片。石蜡切片二甲苯脱蜡 2 次,梯度乙醇水化,微波修复抗原10min,3%双氧水孵育15min 以阻断内源性过氧化物酶,PBS冲洗后5%山羊血清封闭,分别加入鼠抗人α-SMA、OPN、P53、TRIM25、MDM2一抗,稀释比例为1∶100,HRP标记的抗鼠二抗37℃孵育30min;各步骤用PBS洗3min×3次,DAB显色剂,苏木素轻度复染,脱水透明后封片。在显微镜下观察阳性染色区域为棕黄色或棕褐色颗粒;采用Image-Pro Plus 6图像分析软件(美国Media Cybernetics公司)对免疫组织化学染色进行计量分析。从每张切片中连续手动随机取5个高倍视野(×200倍)。分别对切片免疫组织化学染色的阳性区域、阳性细胞数进行定量分析,计算每视野阳性染色的积分吸光度(IOD)值,并取积分吸光度之和。
3.Western blot法实验:将取得的组织在1~2ml匀浆皿中于4℃ 生理盐水中洗净、称量、研磨,加用400μl单去污剂裂解液PMSF,裂解30min后在4℃下12000r/min离心5min,取上清于0.5ml EP管,取少量蛋白提取液,使用PerkinElmer酶标仪测定蛋白含量(波长560nm),标本储存于-80℃备用。将蛋白质样品液与上样缓冲液混合,煮沸5min,SDS-PAGE凝胶电泳分离蛋白,半干转法(北京六一公司)将蛋白质转至PVDF膜,室温下用5%脱脂奶粉封闭2h,加入一抗GAPDH(1∶1000),α-SMA、OPN、P53、MDM2、TRIM25、p-P53(1∶500)。4℃过夜,加带显色剂的抗鼠二抗,室温震动下孵育2h,TBST避光洗涤(5min×3次)后直接奥德赛扫膜仪(美国Licor公司)上扫膜,再对条带进行背景调节和灰度值测定并分析。
4.RT-PCR检测:称取200mg超低温冻存的主动脉中层组织使用液氮碾磨法碾磨成粉末,加入1ml Trizol溶液充分匀浆,将匀浆液转移至EP管中,室温静置5min,向上述匀浆裂解液中加入1/5体积氯仿,充分混合至乳白色,室温静置5min,12000×g离心力 4℃离心15min,取上层无色上清液至另一EP管中,向上清液中加入0.5~1.0倍体积的异丙醇,上下颠倒充分混匀后室温静置10min,12000×g离心力 4℃离心10min,弃上清,加入与Trizol等体积的75%乙醇,轻轻颠倒洗涤,7500×g离心力 4℃离心5min后弃去上清,倒扣EP管干燥沉淀后,加入适量的RNase-free水溶解沉淀,紫外分光光度计检测RNA的 纯度和浓度。按照日本TaKaRa公司反转录试剂盒说明书(RR047A),反应分两步,步骤一:取一PCR管,分别加入5×gDNA Eraser Buffer 2μl、gDNA Eraser 1μl、Total RNA 1μg,然后用RNase Free dH2O补足至10μl,于PCR仪上42℃保温2min,迅速置冰上冷却;步骤二:提前配制Master Mix,将步骤一的10μl反应液与Master Mix 10μl混匀后于PCR仪上按照37℃ 15min→85℃ 5s→4℃∞设定程序进行反应,上述所有反转录程序均为1个循环。反转录完成后按照TaKaRa公司RT-PCR试剂盒(RR820A)进行PCR,反应体系如下(总体积20μl):SYBR Premix Ex Taq 10μl+PCR Forward Primer (10μmol/L) 0.8μl+PCR Reverse Primer (10μmol/L) 0.8μl + ROX Reference Dye Ⅱ0.4μl+DNA模板2μl+dH2O 6μl;扩增程序如下:Stage 1(1 Reps): 95℃ 30s→Stage 2 (40 Reps): 95℃ 5s→60℃ 34s→Dissociation。所有引物均在上海生物工程股份有限公司合成,各基因引物序列如下:P53上游引物:5′-AGGTTGGCTCTGACTGTACC-3′,下游引物:5′-GATTCTCTTCCTCTGTGCGC-3′;MDM2上游引物:5′-CAGGCAGGGGAGAGTGATA-3′,下游引物:5′-GTGATGGAAGGGGGGGATT-3′;TRIM25上游引物:5′-AATACACGGAAATGAAGGCT-3′,下游引物:5′-AACTCATCCCTCTTGGTCAG-3′;β-actin 上游引物:5′-AGCGAGCATCCCCCAAAGTT-3′,下游引物:5′-GGGCACGAAGGCTCATCATT-3′。

结 果
1.免疫组化检测主动脉中层平滑肌细胞变化:主动脉中层的主要细胞成分就是平滑肌细胞,免疫组化证实获取组织主要细胞成分是α-SMA阳性的平滑肌细胞。已有研究表明VSMC由分化型转变成未分化型会导致SMC收缩功能减弱,而这有可能导致主动脉中层组织薄弱。在本实验中,分别检测主动脉中层分化型VSMC表型标志物α-SMA和未分化型表型标志物OPN。通过免疫组化染色实验发现,与正常对照组比较,在AD组血管中层的α-SMA含量显著降低而OPN含量显著升高,差异有统计学意义(P<0.01,图1,表2)。

图1 两组主动脉α-SMA和OPN表达免疫组化(×200)与正常对照组比较,*P<0.01

表2 主动脉α-SMA和OPN免疫组化IOD值
2.免疫组化检测主动脉中层P53、MDM2和TRIM25含量变化:通过免疫组化检测发现AD组中层的P53、MDM2和TRIM25含量与正常对照组比较显著增多,而p-P53蛋白表达量下降比较差异有统计学意义(P<0.01,图2,表3)。
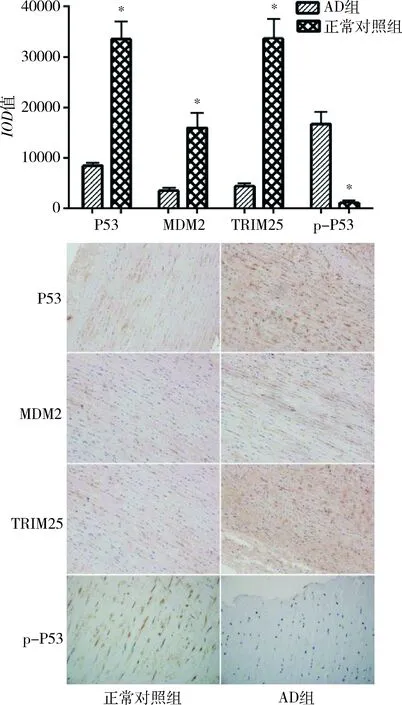
图2 AD组和正常对照组主动脉中层(免疫组化,×200)与正常对照组比较,*P<0.01

表3 P53、MDM2、TRIM25和p-P53 IOD值
3.Western blot法检测主动脉中层蛋白表达:与正常对照组比较,α-SMA和p-P53在AD患者主动脉组织内表达显著降低(P<0.01),而在AD患者主动脉组织中OPN、P53、MDM2、TRIM25蛋白水平却显著增高,差异有统计学意义(P<0.01,表4,图3)。
4.RT-PCR检测结果:进一步通过RT-PCR检测了P53、MDM2和TRIM25的mRNA表达,发现夹层组中P53、MDM2和TRIM25的mRNA表达量也增加(P<0.01,表5,图4)。
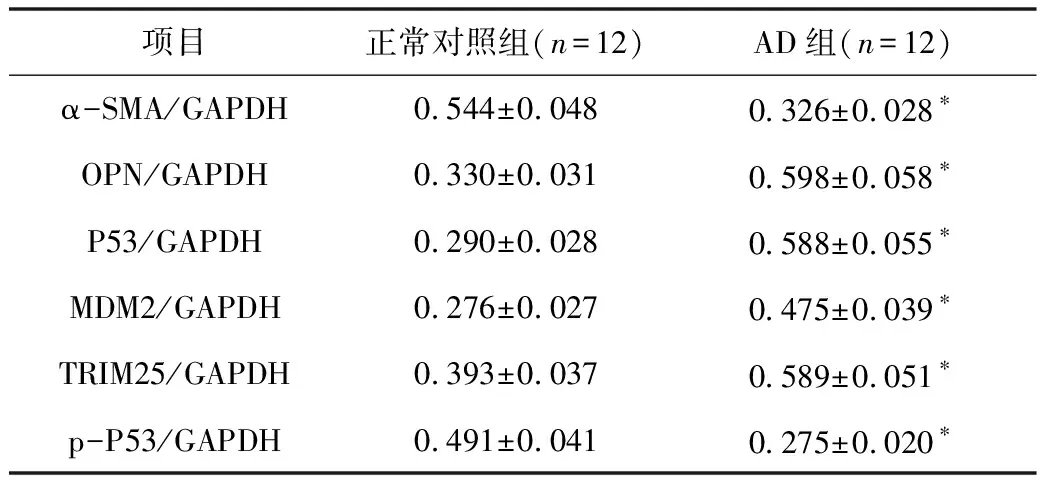
表4 AD组和正常对照组相关蛋白表达水平
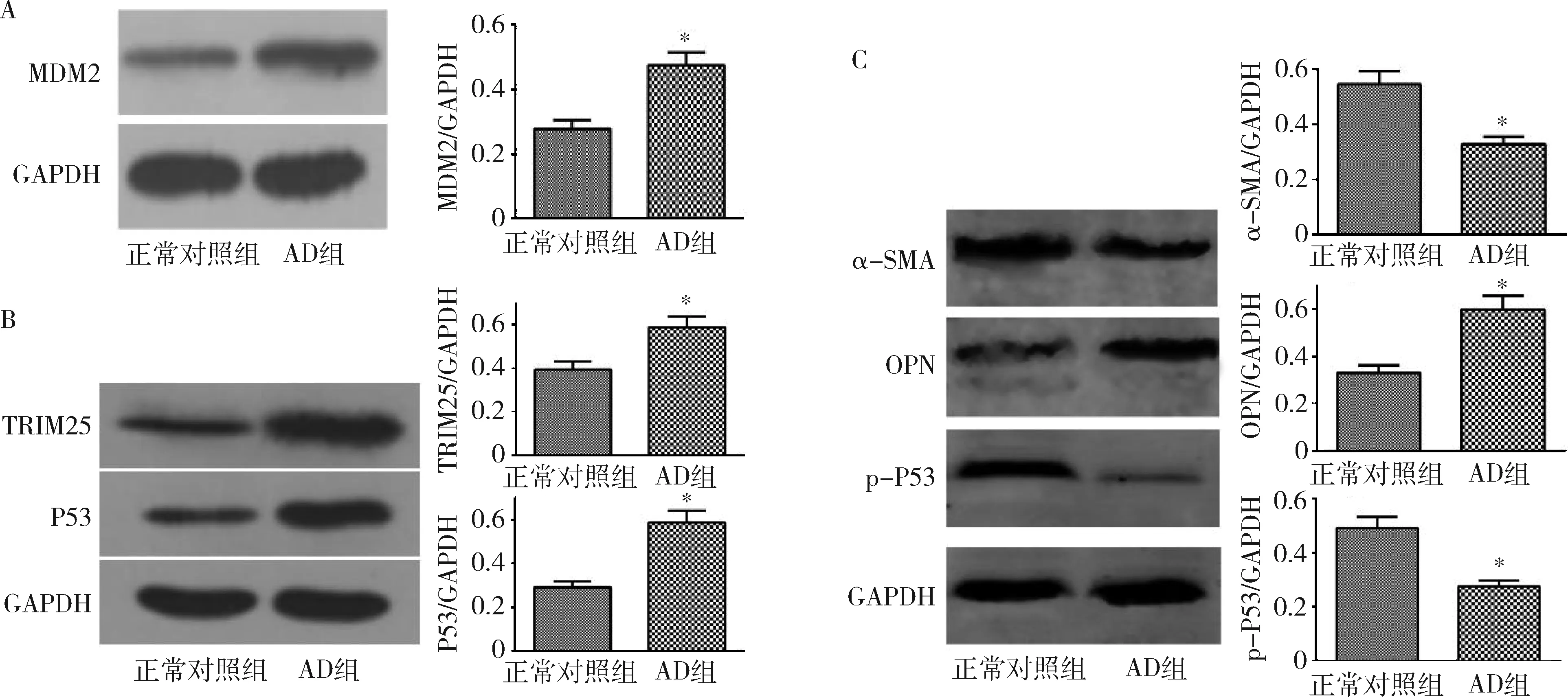
图3 Western blot法检测结果A.MDM2;B.TRIM25和P53;C.α-SMA、OPN和p-P53;与正常对照组比较,*P<0.01

mRNA正常对照组(n=12)AD组(n=12)P531.000±0.0002.235±0.203∗MDM21.000±0.0001.697±0.203∗TRIM251.000±0.0001.794±0.191∗
与正常对照组比较,*P<0.01
讨 论
AD的典型组织学特征是中层退行性变,目前对主动脉中层退行性变的分子机制正被广泛研究,但尚未阐明[6]。大量研究显示微纤维、胶原、蛋白聚糖和黏多糖、血管VSMC等主动脉壁的组成结构和功能异常是AD发生的病理基础。而VSMC的表型转化和凋亡在AD发生、发展中扮演重要角色,其受多种信号通路调节包括MAPK、PI3K和cAMP等[7,8]。最近发现P53 是 VSMC 分化的强诱导因子,P53 导致VSMC由去分化型向分化型转化,分化型VSMC可以维持血管弹性和收缩血管,不易导致主动脉中层退行性变[9]。Leeper等也发现,CDKN2B缺失的情况下,P53 表达上调,动物模型主动脉瘤形成。在本实验中也证实AD组中P53含量明显增多,α-SMA含量减少,说明AD中VSMC由分化型向去分化型转变。但这违背了 P53 作为VSMC 分化诱导因子的功能。

图4 两组P53、MDM2、TRIM25 mRNA表达水平与正常对照组比较,*P<0.01
P53下游转录激活产物之一是MDM2,它可以通过与P53结合形成复合物,维持低水平的P53蛋白表达进而抑制P53介导的转录活性,这个P53/MDM2反馈回路也是保持机体生理功能的重要机制之一。 如果出现了各种刺激因素包括低氧、DNA损伤和核苷酸缺失等情况会导致P53蛋白表达和转录活性迅速增加,从而激活下游的靶基因MDM2。当DNA修复后,负调节因子MDM2开始反馈抑制P53,从而防止P53的进一步作用[10,11]。而在本研究中发现AD组的MDM2含量较正常组显著增加。在AD组主动脉中层组织中,Western blot法检测MDM2的蛋白含量增加,并且通过 RT-PCR检测笔者发现MDM2的mRNA含量较对照组增加数倍。所以,笔者推测在AD发病过程中,MDM2和P53的负反馈平衡被打乱:增加的P53促进MDM2的mRNA表达量增加,使 MDM2的mRNA表达量增多进而增加MDM2的蛋白含量,但是增加的MDM2蛋白不能降解P53蛋白,导致P53蛋白的稳定性增加并致使P53在AD中层组织中聚集。
MDM2可通过多种分子机制参与对P53的反馈抑制,包括:①P53的活性由多种因素决定,激活的P53在细胞核内激活其靶基因的转录,其降解在胞质中进行。核输出序列(NES)和核定位序列(NLS)是MDM2具有核质穿梭能力的重要标志,亚细胞定位对于其抑制P53转录活性非常重要。如果P53在高表达状态,细胞核中的P53可以被MDM2直接降解。如果P53在低表达状态时,其转录激活位点可以被具有MDM2突变的NES结合,从而抑制P53活性; ②MDM2的N端具有疏水和芳香氨基酸形成的深沟,P53作为转录因子TFIID的组成部分与MDM2结合后抑制了其转录激活靶基因的能力; ③P53蛋白的特异性E3连接酶就是MDM2,其可以使P53蛋白和泛素相连接后泛素化P53,再被蛋白酶在细胞质中降解[12,13]。
而TRIM25作为一种泛素化E3连接酶,主要表达在胚胎、子宫、甲状腺、主动脉和脾脏中表达,很少在其他组织中表达。有研究报道,在经雌激素治疗过的乳腺癌患者中可检测到高水平的TRIM25、P53和MDM2的表达[14]。而本研究发现夹层组的TRIM-25蛋白和mRNA水平的表达量均显著增加。因而笔者推测可能由于夹层中TRIM-25的表达量增加竞争性抑制MDM2与P53的泛素化降解从而增加P53含量。但是P53的活性受到抑制,从而不能诱导VSMC分化,通过Western blot法检测也发现夹层组P53的磷酸化水平降低。
综上所述,AD主动脉中层中P53、MDM2及TRIM 25表达量均显著增加,主动脉中层中VSMC表型向去分化型转变,可能是由于P53/MDM2反馈环平衡被TRIM25打乱所致。但是P53、MDM2及TRIM25在主动脉中层组织中的增多究竟是AD发病之前的诱因或者是其发病之后的结果,同时高血压对血管平滑肌细胞的影响如何?这需要进一步在体外细胞和动物实验来验证TRIM-25的过表达是否会导致MDM2和P53结合障碍从而引起其含量增加,导致VSMC表型和功能发生变化。
