Interaction of TATB with Cu and Cu+1.A DFT study
2019-03-01Lemirker
Lemi Türker
Middle East Technical University,Department of Chemistry, Üniversiteler,Eskis¸ehir Yolu No:1,06800 Çankaya/Ankara,Turkey
Keywords:Triaminitrinitrobenzene TATB Tautomerism Copper DFT Pull-push
A B S T R A C T Symmetric triaminotrinitrobenzene known as TATB decomposes in contact with some heavy metals,such as copper.Presently,TATB and some of its important tautomers are subjected to B3LYP/6-31G(d,p)level of treatment.Some molecular orbital energies have been obtained and compared.Then,interaction of these species with Cu and Cu+are investigated.Although TATB has been found to be affected little by either Cu or Cu+,undergoing only some bond length changes and/or distortions,the resonance-assisted tautomers having one to three aci groups decompose by the in fluence of Cu atom.In each case,only one N-OH bond rupture occurs.
1.Introduction
The thermal stability is one of the most important properties of a high explosive(HE)effective on its practicality when employed as a constituent of either conventional or nuclear weapons.Regarding to the formulation,processing,and handling of an HE,thermal stability is of main concern but also important for its safety including fuel fires,propellant fires,and even a potential for sympathetic detonation.An increased emphasis on such hazards has renewed interest in understanding the fundamental relationship between molecular structure and reactivity of energetic materials.
It has been long known that 1,3,5-triamino-2,4,6-trinitrobenzene(TATB)is a reasonably powerful high explosive having thermal and shock stability which is considerably greater than that of any other known material of comparable energy.Consequently,it is a very attractive candidate for the main HE charge in many weapons.Desirable properties of TATB has made it the subject of numerous studies.
The first mention of TATB in the literature goes back to 1888.It was obtained by Jacson and Wing by treating 1,3,5-tribromo-2,4,6-trinitrobenzene with cold alcoholic solution of ammonia[1,2].It decomposes at 360°C without melting[3].It is a yellow crystalline solid that is insoluble in most solvents.The best solvent is hot concentrated sulfuric acid.After II world war,research efforts focused on high energetic materials for safer and more heat resistant explosives[4].
The compound TATB has been known for its unusual thermal properties since the 1950s.A striking feature of TATB is the strong and extensive hydrogen bonding,both inter-and intramolecular,between NO2and NH2groups[5,6].The first important work on the reason for this unique stability was done in 1965 b y Cady and Lar s on,who determined the crystal structure of TATB[5].Since then intense research has been done on TATB[7-18].Manaa and Fried reported the results of second-order Møller-Plesset(MP2)and density functional theory(DFT)calculations of the rotational barrier and torsional potential for intramolecular rotation of the nitro group in TATB[19].Geometrical parameters,aromaticity,and conformational flexibility of the set of polysubstituted benzenes(including TATB)with different number and position of nitro and amino groups were calculated theoretically[20].Zhang et al.considered TATB/Graphene sandwich type complex computationally[21].Synthetic TATB processes quantum-chemically were investigated by Radhakrishnan et al.using density functional theory(DFT)[22].DFT was employed in order to study the thermodynamic parameters of TATB and its fullerene derivatives[23].
In the present study,some important tautomeric forms of TATB and interaction of them with some copper species have been investigated within the realm of DFT.
2.Method of calculations
In the present treatise,the geometry optimizations were performed on conformationally stable(MM2 molecular mechanics calculations)forms.Then the optimizations were achieved by PM3,HF and then density functional theory(DFT)[24]calculations using B3LYP hybrid exchange-correlation potential[25].For closed shell systems B3LYP/6-31G(d,p)and open-shell systems UB3LYP/6-31++G(d,p)[26]type calculations were performed.Note that 6-31++G(d,p)basis set is available for copper[26,27].The exchange term of B3LYP consists of hybrid Hartree-Fock and local spin density(LSD)exchange functions with Becke's gradient correlation to LSD exchange[28].The correlation term of B3LYP consists of the Vosko,Wilk,Nusair(VWN3)local correlation functional[29]and Lee,Yang,Parr(LYP)correlation correction functional[30].The BLYP method gives a better improvement over the SCF-HF results.The optimizations in the present study have been obtained by the use of SPARTAN 06 program[31].
3.Results and discussion
TATB molecule,having alternatingly substituted nitro and amino groups should allow existence of push-pull type interaction.Below some contemplation on the structure of TATB has been presented.
3.1.Structure of TATB
Manaa and Fried by studying the rotational barrier and torsional potential for intramolecular rotation of the nitro groups in TATB found that the calculated rotational barrier two to three times higher than that of nitrobenzene and nitroethylene[19].They attributed this result to very strong intramolecular hydrogen bonding in this molecule.Their MP2 optimization resulted in a nonplanar structure,while the B3LYP produced a planar one similar to the experimentally observed structure in the crystalline form.Omelchenko et al.,studied geometrical parameters,aromaticity,and conformational flexibility of the set of polysubstituted benzenes with different number and position of nitro and amino groups[20]and concluded that the presence of nitro and amino groups in vicinal positions formed strong intramolecular resonance-assisted hydrogen bonds.Also they studied TATB molecule,which possesses alternatingly substituted nitro and amino groups and should have strong intramolecular resonanceassisted hydrogen bonds.
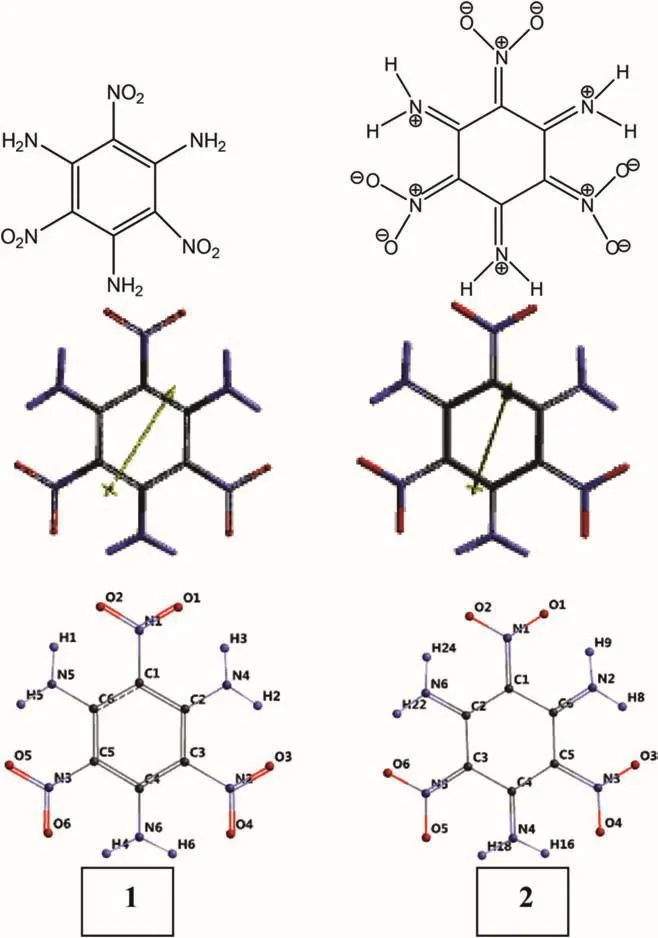
Fig.1.Two optimized resonance forms of TATB(B3LYP/6-31G(d,p))and their numbering.
Fig.1 shows two canonical structures of TATB and their numbering,together with optimized structures such that in the shown form 1 and 2 possess aromatic benzene ring and notaromatic hexagonal ring,respectively.In the later case the nitro and amino substituents have exocyclic carbon-nitrogen double bonds.Structure 2 is an interesting highly charge separated resonance form obtained from 1.
Supplementary tables,Table S1 and S2 show the coordinates of structures 1 and 2,respectively.Note that respective atoms in those structures not only have different bonding topology but also have different coordinates.Tables S3 and S4 display the Mulliken bond order matrices for those structures.
Fig.2 displays some tautomeric forms of TATB molecule.Tautomerism in TATB destroys the aromatic nature of it.So,none of the structures in Fig.2 is aromatic.Note that of the various possible tautomeric forms,Fig.2 includes one(3),two(4)and three acigroup having structures of which 5 and 6 stand for an asymmetric and symmetric triply tautomeric forms,respectively.
All the tautomers shown in the figure are 1,5-proton tautomers which are in general less likely to occur as compared to 1,3-tautomers,however presence of some special structural conditions may stabilize them.Also note that 1,3-tautomerism is not suitable for TATB molecule.
Fig.3 stands for bond lengths of TATB in aromatic(1)and notaromatic(2)forms.As seen in the figure structures seem to be equivalent(any specific bond lengths calculated are equal in these structures up to two significant figures).
Apparently identical calculated IR-spectra have been obtained for structures 1 and 2(Fig.4)although frequencies are slightly different.Most of the modes are coupled with each other yielding apparently identical spectra.Note that one of the N-H stretchings(symmetric)has lower amplitude compared to the other(asymmetric).
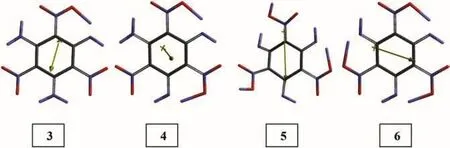
Fig.2.Some tautomeric forms of TATB(B3LYP/6-31G(d,p)).

Fig.3.Bond lengths of TATB in aromatic(1)and not-aromatic(2)forms(B3LYP-6-31G(d,p)).

Fig.4.IR spectra of TATB structures 1 and 2(B3LYP-6-31G(d,p)).

Table 1 Various energies of TATB species(B3LYP/6-31G(d,p)).
Various energies of TATB species are shown in Table 1.The stability of structures follow the order of 1> 2>3> 6>4> 5.Also B3LYP/6-31++G(d,p)level of calculations predict 1 to be more stable than 2 but comparable.Note that structure 2-6 are all notaromatic.Structures 1 and 2 could be considered as resonance forms because they are converted to each other via series of electron shifts.However they have different coordinates(See Tables S1 and S2).However,push-pull interactions operative between amino and nitro groups in the proximity(1,2-positions)or through 1,4-positions causes structures 1 and 2 to be closely related.The energy data of structures 1-6 are shown in Table 1 where ZPE,E and Ecare zeropoint vibrational energy,total electronic energy and the corrected total electronic energy values,respectively.The table indicates that the contribution of structure 2 into the resonance hybrid of TATB is almost as high as structure 1.Structure 2 may form various resonance-assisted tautomeric forms 3-6.Omelchenko et al.employing various aromaticity indices concluded that structure 2 has very low aromatic character[20].Although it is not aromatic,it is almost as stable as structure 1.They related this fact with hydrogen bonding between adjacent NH2and NO2groups assisted by push-pull effect.The total energy of hydrogen bonding was found to be very high.As evident from the order of stabilities,the tautomeric forms are less stable than either of 1 or 2.Asymmetric triply tautomeric form(5)is the least stable due to improper conformational form and highly charge separated character of it.The PM3 calculations over B3LYP/6-31G(d,p)optimized structures indicate that all of them are endothermic.Note that 5 is the most endothermic structure.Tautomers 3,4 and 6 are less endothermic than both 1 and 2.Note also that the least stable structure is 5 and its heat of formation value far exceeds the others because,as mentioned above,it has two aci-NO2groups whose conformations are not preferable for any suitable hydrogen bonding.Structure 1 is the most stable and mostendothermic one in Table 1.Although,in 2 aromaticity is lost,the push-pull type interaction and consequent favorable charge-charge interactions through space stabilize it.
Fig.5 shows the HOMO,LUMO,HOMO-1(NEXTHOMO)and LUMO+1(NEXTLUMO)patterns of two forms of TATB(1 and 2 are aromatic and not-aromatic,respectively).Although their HOMO-1,HOMO and LUMO+1,patterns are different,they have identical LUMO pattern.
Fig.6 shows the distribution of some molecular orbital energy levels of structures 1 and 2.Although they appear to be the same in Fig.6,there exist some energy differences.See Table 2 for the HOMO,LUMO energies and interfrontier energy gaps(Δε=εLUMO-εHOMO).

Fig.5.HOMO,LUMO,HOMO-1 and LUMO+1 patterns of two forms of TATB(B3LYP-6-31G(d,p)).
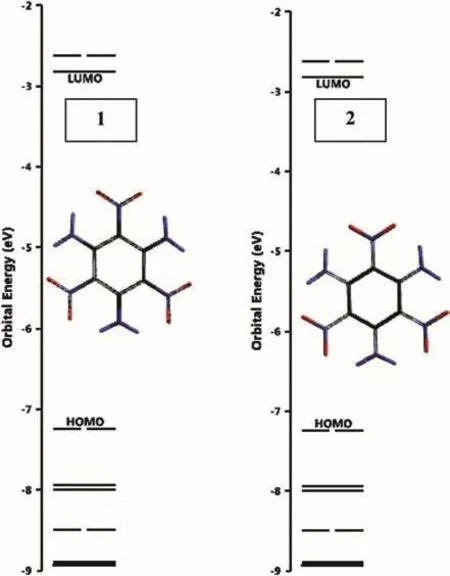
Fig.6.Some molecular orbital energies of structures 1 and 2(B3LYP-6-31G(d,p)).
As seen in Fig.6 and Table 2,Δε values of 1 and 2 are very comparable with each other.Therefore,their UV-VIS spectra are almost the same(Fig.7).The conjugated paths in 2 should be short because it does not have an aromatic core for the extended conjugation.However,inspection of the HOMO patterns of 1 and 2 indicates that not only 2 but also structure 1 possesses short conjugation paths embedded.Thus,they have very similar UV-VIS spectra absorbing in the UV region.The Δε values follow the order of 5<4<1<6<2<3.
3.2.Interaction with copper species
As indicated in the above section structure 2 has great resemblance to structure 1 in terms of geometrical properties and energetics.It has been reported that TATB in contact with heavy metals such as copper undergoes decomposition[3].Figs.8 and 9 show the optimized structures of some TATB+Cu,TATB+Cu+species(7-10)and their bond lengths,respectively.Note that notation,TATB+Cu,used here denotes the composite system but not cuprous ion.Also note that structures 7-10 originally related to structure 1 which has an initially an aromatic nucleus.Also note that calculations for the open-shell systems the level of calculations are UB3LYP/6-31++G(d,p).From the figures it is evident that the copper atom and cuprous ion do not affect much structures 1 and 2,except some distortions and bond length changes(no bond cleavages occur!).
From Figs.8 and 9 it is observed that Cu atom in 7 and 8 are in between NH2and NO2moieties.Whereas in 9 and 10,Cu+ion is nearby the NH2substituent but in the vicinity of NO2in 10.Note that 9 and 10 are originated from 1 and 2,respectively.Although,1 and 2 have many common characteristics,by the presence of copper atom or cuprous ion differential behavior arises.
Figs.10 and 11 show the electrostatic charges(ESP)on the systems of present concern.The ESP charges are obtained by the program based on a numerical method that generates charges that reproduce the electrostatic potential field from the entire wavefunction[31]From Fig.10 one realizes that structures 1 and 2,resembling each other from many aspects some what differ in terms of charge distribution.In each case copper atom donates some electron population to organic molecule acquiring itself some positive charge.
Fig.12 shows the frontier molecular orbital paterns of TATB+Cu composites 7 and 8 originating from structures 1 and 2,respectively.Note that UB3LYP/6-31++G(d,p)level of calculations have been performed(because of unpaired electron of copper atom)thus αandβtype orbitals arise.As seen in the figure,the copper atom has great contribution to HOMO orbitals both in 7 and 8.Whereas,some minute contribution to LUMO orbitals occurs.In the case of 7 α-HOMO spreads over the oxygens of the nearby nitro group andthe copper atom.Theβ-HOMO covers the ortho amino group,the above mentioned nitro group and the copper atom.A similar pattern occurs in the case of structure 8.
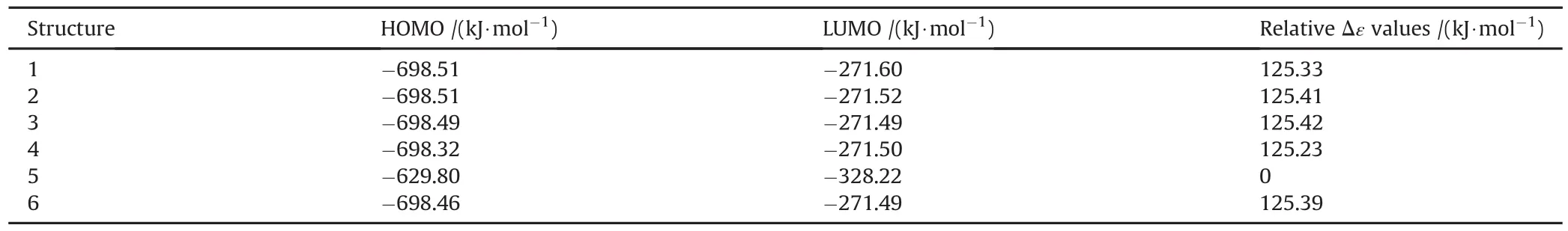
Table 2 The HOMO,LUMO energies and interfrontier energy gaps(Δε)of TATB species.

Fig.7.UV-VIS spectra of structures 1 and 2(B3LYP/6-31G(d,p)).
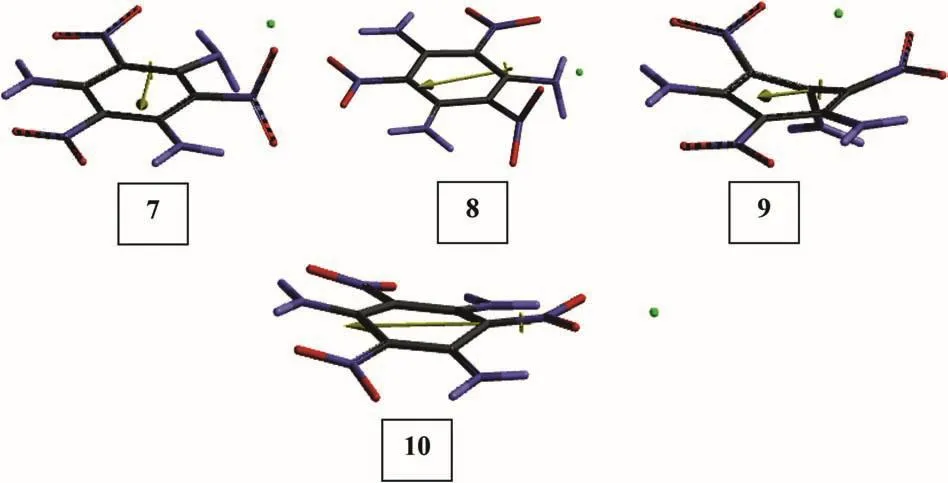
Fig.8.Optimized structures of some TATB+Cu(7,8)(UB3LYP/6-31++G(d,p))and TATB+Cu+(9,10)(B3LYP/6-31++G(d,p))species.

Fig.9.Bond lengths of optimized structures of some TATB+Cu(7,8)(UB3LYP/6-31++G(d,p))and TATB+Cu+(9,10)(B3LYP/6-31++G(d,p))species(hydrogens not shown).
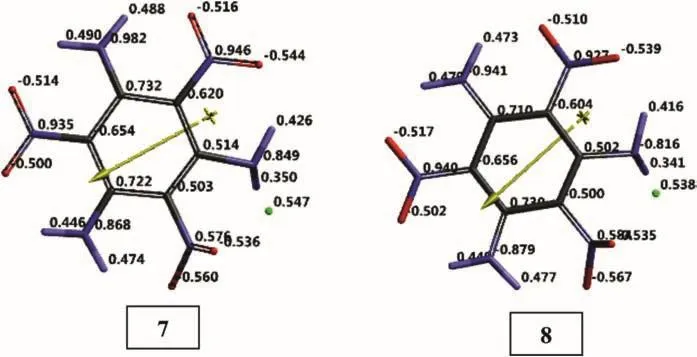
Fig.10.Electrostatic charges(ESP)on TATB+Cu species(UB3LYP/6-31++G(d,p)).
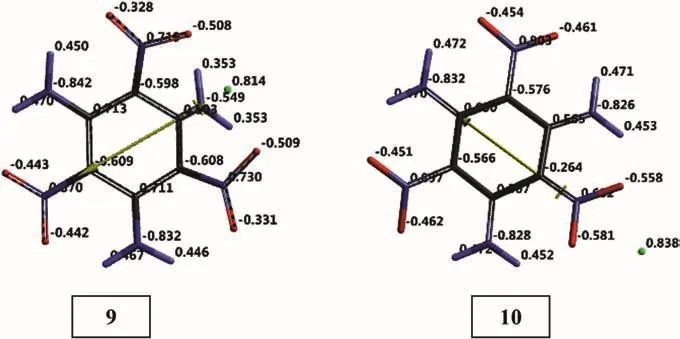
Fig.11.Electrostatic charges(ESP)on TATB+Cu+species(B3LYP/6-31++G(d,p)).
Fig.12 shows that cuprous cation gets some electron population from the organic molecule and its initial positive charge decreases.So,Cu+acts as an oxidizing agent.Note that the position of Cu+ion,that is nearby the NH2moiety in the case of structure 9(originates from 1)but next to the NO2in the case of 10(originates from 2).
Effectof cupric ion on 1 and 2 has been not considered presently,because copper atom in the neutral state has striking effect on the tautomers to decompose them prior to formation of cuprous and cupric ions.Due to similar type of reasoning,the effect of cuprous ion on the tautomers has not been investigated.On the other hand,structure 5(one of the tautomers of TATB)is the least stable tautomer.It formed by the transfer of proton from the inconvenient site of NH2group to NO2.Therefore,it was not considered for Cu containing species.
Fig.13 reveals that in the case of tautomeric TATB species,the copper atom has tendency to donate some electron population and aci N-OH bond breaks down to generate OH-ion.So,TATB tautomers act as oxidizing agent.
Figs.14 and 15 display the optimized structures of some tautomeric TATB+Cu species and their bond lengths,respectively.It is evident from the figures that tautomeric forms are not stable in the presence of Cu.Since,copper atom causes their decomposition,interaction of them with Cu+has been ignored because normally Cu+accompanies copper or Cu2+,having intermediate oxidation state between them.
The ruptured bond in the tautomers of interest is the aci N-OH linkage and only one of them is affected if there exist more then one of them.
Table 3 tabulates various energies of the Cu and Cu+containing composite structures presently concerned.Structures 7 and 8 which are originated from 1 and 2,respectively have comparable energies,7 being more stable one.Structure 9 originating from 1 is favored energetically over 10 which is related to structure 2.The stability order is 11>13>12>7>8>9>10.
As for the copper composites of tautomeric forms(11-13),they have the stability order of 11>13>12.Since,after the bond cleavage,the remaining organic moiety has positive charge,the stabilization of it depends on the position and kind of the substituents and the extent of hydrogen bonding present in the tautomeric structures.Thus the above stability order arises.
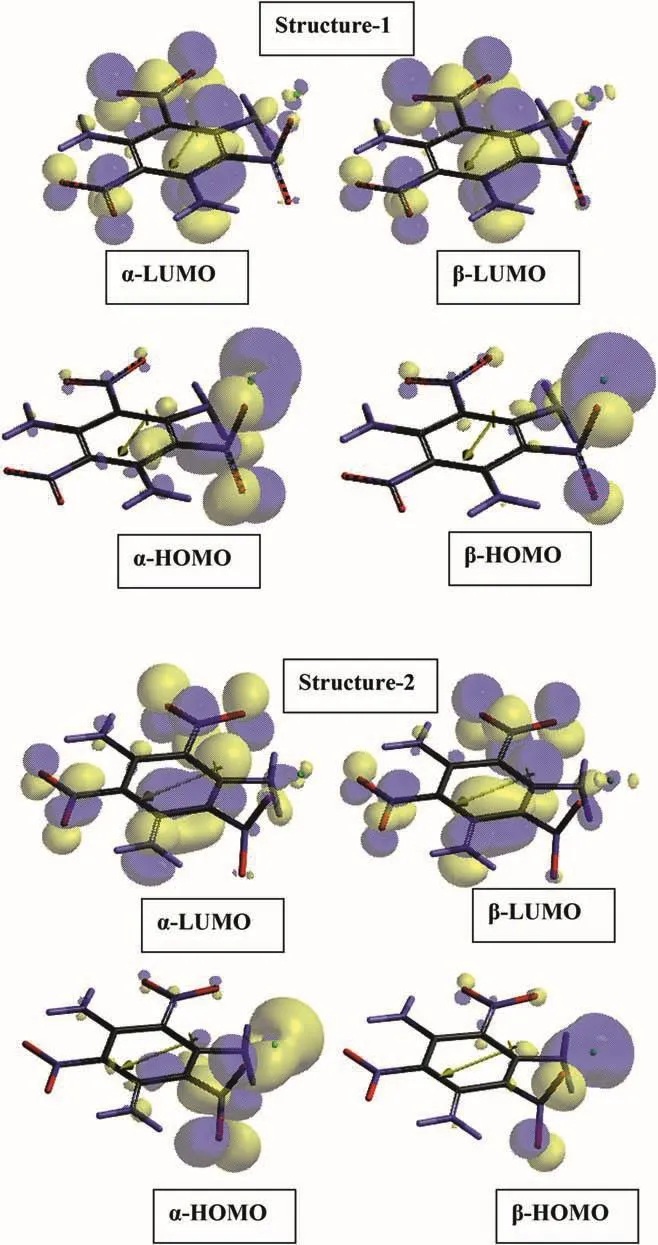
Fig.12.Frontier molecular orbital paterns of TATB+Cu composites structures 7 and 8(UB3LYP/6-31++G(d,p)).
Considering TATB-originated structures as host(H),Cu or Cu+as guest(G)and the composite structures(H°G)(as suggested in general by Anslyn and Dougherty[32])binding energies have been calculated.The relative binding energies for structures 7-13 are shown in Table 4 and the order is 13>11>9>10>12>7>8 within the constraints of the host-guest model.The order indicates that not-aromatic structure 2,has greater interaction with Cu or Cu+(to form 9 and 10)than structure 1(to form 7 and 8)because 2,beside the other factors,is more charge-separated structure.However,in each case,the interaction with Cu+is less than it has been with Cu.The tautomeric structures(11-13.)which have progressively increasing number of tautomeric sites are characterized with the order of 13>11>12.
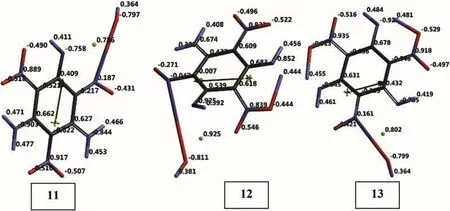
Fig.13.Electrostatic charges(ESP)on tautomeric TATB+Cu species(UB3LYP/6-31++G(d,p)).
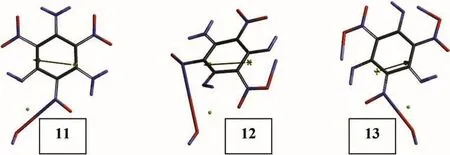
Fig.14.Optimized structures of some tautomeric TATB+Cu species(UB3LYP/6-31++G(d,p)).
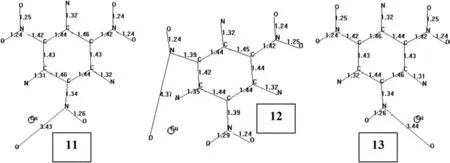
Fig.15.Bond lengths of optimized structures of some tautomeric TATB+Cu(UB3LYP/6-31++G(d,p))species(hydrogens not shown).
Fig.16 shows some of the molecular orbital energy distribution of tautomeric TATB+Cu species(11-13)compared to 7.Note that 7 originates from 1 and the others are tautomeric forms.Also note that all the structure in Fig.16 are isomeric Cu composites.Since Cu atom has an unpaired electron,the calculations have been performed at the unrestricted level(UB3LYP/6-31++G(d,p)).
Table 5 displays the HOMO,LUMO energies and the interfrontier molecular orbital energy gaps(Δε)for Cu and Cu+composites of TATB species.As evident from Table 5,the HOMO and LUMO energies of 8 are lower than the respective values of 7.The HOMO and LUMO energies of 9 is lower then 10 butΔε of 10 is greater than 9,because Cu+ion unequally affects the HOMO and LUMO energies so Δε order is reversed.The number of aci groups and their positions are in fluential on the extentof conjugations.So,the aci groups raise or lower the HOMO and LUMO energies and consequently dictate Δε values.
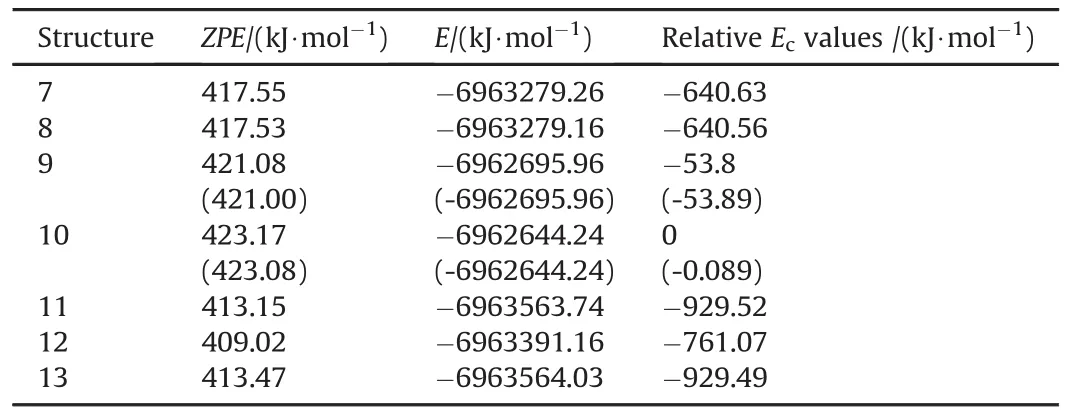
Table 3 Various energies of some TATB+Cu species.
4.Conclusion
The present study puts some light on to some quantum chemical properties of sym-triamminotrinitrobenzene which is an important insensitive energetic material.Density functional treatment reveals that Cu and Cu+species interact with sym-TATB without any bond cleavage.However,the resonance-assisted tautomers of it are destructively affected by Cu atom undergoing rapture of only one N-OH linkage of the aci form.Therefore,in preparation of certain ammunition with TATB,the other components/additives should not induce or shift the tautomeric equilibria to the side of resonance-assisted tautomers.

Table 4 Relative binding energies of Cu or Cu+containing TATB-originated structures.

Fig.16.Effect of copper on some of orbital energies of TATB and its tautomers presently considered(UB3LYP/6-31++G(d,p)).
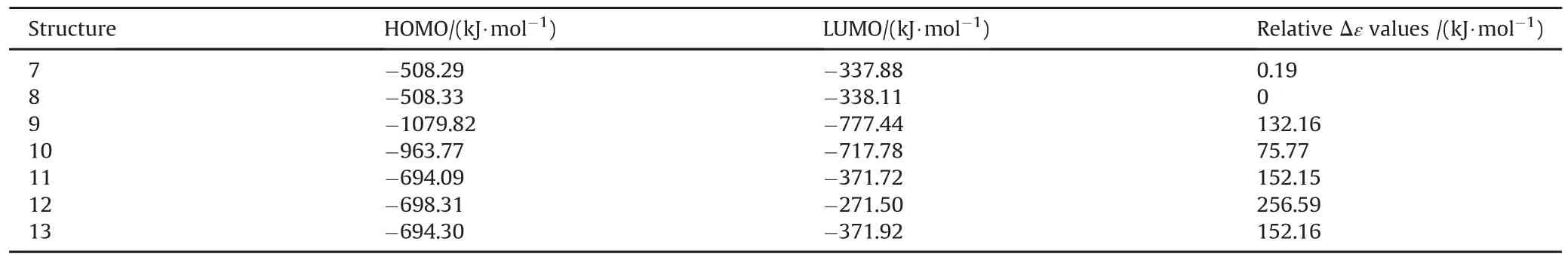
Table 5 The HOMO,LUMO energies and interfrontier energy gaps of some TATB+Cu species.
Appendix A.Supplementary data
Supplementary data related to this article can be found at https://doi.org/10.1016/j.dt.2018.05.001.
杂志排行

Defence Technology的其它文章
- Fluorine-containing oxidizers for metal fuels in energetic formulations
- On the formation of Basu's Type III(peeled orange)gunshot residues
- Control of exterior ballistic properties of spin-stabilized bullet by optimizing internal mass distribution
- Crystal lattice free volume and thermal decomposition of nitramines
- Tribological and vibrational characteristics of AISI 316L tested at elevated temperature and 600 Torr vacuum
- Underwater explosion effects of 60 mm H.E.mortar bomb on a cylindrical concrete structure-PIT