小RNA病毒3C蛋白功能的研究进展
2019-02-28曹圣慧黄孝天
曹圣慧,黄孝天
1 小RNA病毒科的基因组结构与致病性
1.1小RNA病毒基因组结构 小RNA病毒科基因组为单正链RNA,大小为6.7~10.1 kb,病毒颗粒呈球形,无被膜包被,病毒衣壳为二十面体结构[1]。小RNA病毒基因组结构高度保守,包括5′非编码区(5′-noncoding region,5′-NCR)、开放阅读框、3′非编码区(3′-NCR)和3′端的polyA尾。5′-NCR包含多个RNA二级结构元件,其中一个重要的二级结构元件是内部核糖体进入位点(Internal ribosome entry site,IRES),IRES三叶草结构与细胞蛋白质相互作用后招募核糖体,启动病毒基因组的翻译。病毒基因组开放阅读框包括3个部分,分别为P1区、P2区和P3区。P1区经翻译加工后形成结构蛋白VP1、VP2、VP3和VP4,组成病毒的衣壳结构。P2区和P3区分别翻译加工形成非结构蛋白2A、2B、2C以及3A、3B、3C、3D。其中,2A蛋白和3C蛋白分别参与病毒蛋白的剪切。3′-NCR和病毒的复制效率相关,3′端的polyA尾参与病毒的复制和翻译[2]。
1.2常见小RNA病毒的致病性 小RNA病毒科包括35个属,80个种[1]。许多小RNA病毒可引起人和动物的脑部、心脏、肝脏、皮肤、胃肠道或上呼吸道疾病,且目前无有效的疫苗用于治疗。常见的小RNA病毒包括肠道病毒71型(Enterovirus 71,EV-71)、肠道病毒D68型(Enterovirus D68,EV-D68)、柯萨奇病毒(Coxsackievirus,CV)、脊髓灰质炎病毒(Poliovirus,PV)、甲型肝炎病毒(Hepatitis A virus,HAV)、鼻病毒(Human rhinovirus,HRV)、口蹄疫病毒(Foot-and-mouth disease virus,FMDV)、脑心肌炎病毒(Encephalomyocarditis virus,EMCV)等。EV-71、CV-A2、CV-A16、CV-A6和CV-A10可导致每年亚太国家数百万儿童患手足口病,甚至引起更严重的临床症状,如无菌性脑膜炎、急性迟缓性麻痹和神经呼吸系统综合征[3]。CV-B4可引起新生儿心肌炎、Ⅰ型糖尿病以及神经性疾病[4]。EV-D68可引起世界范围内人类呼吸道疾病的流行[5]。EMCV可引起动物脑炎、心肌炎为主要特征的急性传染病,FMDV可导致牛羊等动物口蹄疫的暴发流行,对农业生产造成重大的经济损失[6-7]。
2 小RNA病毒3C蛋白的结构
小RNA病毒的3C蛋白包含175~253个氨基酸,大小为19~28 kD。3C蛋白集丝氨酸蛋白酶和半胱氨酸蛋白酶特性为一体。作为丝氨酸蛋白酶,3C蛋白具有催化三联体Cys-His-Glu/Asp[8]。作为半胱氨酸蛋白酶,3C蛋白具有特征催化基序Gly-X-Cys-Gly[9]。近年来,多种小RNA病毒如HRV、PV、HAV等的3C蛋白晶体结构显示,3C蛋白都具有二个相同的、6条反向平行链构成的、呈约90°的β-桶状(β-barrel)结构域,二个结构域之间延伸出一个可结合底物的浅槽。其中,由十几个氨基酸残基构成的环状结构位于浅槽的上方,称为β-折叠(β-ribbon),它对底物的特异性识别具有重要作用[9](图1)。研究显示HRV、PV、HAV、FMDV中3C蛋白的β-折叠为闭合构象,而EV-71中3C蛋白的β-折叠是一种开放构象,位于β-折叠基底的Gly-123和 His-133控制着β-折叠的灵活性[10]。

图1 EV-71的3C蛋白结构[10]Fig.1 Structure of EV-71 3Cpro[10]
3 小RNA病毒3C蛋白的功能
近年来对于小RNA病毒3C蛋白的研究表明,该蛋白参与病毒前体蛋白的剪切,与促进病毒复制、调控细胞凋亡以及逃避免疫应答等密切相关。
3.1促进病毒复制 由于自身基因组的限制,许多病毒依赖于宿主细胞来完成自身的复制。研究发现3C蛋白可裂解多种蛋白如poly-A结合蛋白(poly(A)-binding protein,PABP)、剪接因子脯氨酸和谷氨酰胺(Splicing factor proline and glutamine rich,SFPQ)、蛋白激酶R (protein kinase R,PKR)等。这些蛋白对病毒复制的影响是不同的。Sun等研究表明鸭甲肝病毒(duck hepatitis A virus,DHAV)的3C蛋白可裂解PABP蛋白中 Q367和G368之间的特异性位点,PABP蛋白被裂解为N端片段和C端片段,N端片段的存在利于DHAV的复制,而C端片段则截然相反。C端片段的功能被细胞所抑制的机制尚不清楚[11]。Kobayashi等研究结果显示EMCV的3C蛋白可裂解PABP蛋白中的Q437和G438之间的特异性位点,导致一个N端45 kD的蛋白在病毒感染的细胞中累积,促进病毒的复制。而当PABP的裂解受到干扰时,病毒的复制明显受到抑制。因此,EMCV的3C蛋白可通过裂解PABP而促进病毒复制[12]。此外,Dylan等研究显示HRV的3C/3CD 可裂解SFPQ,而SFPQ水解片段的产生导致病毒复制以及病毒颗粒的增多[13]。Chang等人发现EV-71感染宿主时,3C通过裂解PKR增强了病毒的复制[14]。Yao等发现槲皮素能够结合EV-71 3C蛋白的底物识别位点从而抑制其活性,以及抑制宿主体内的病毒复制,推断槲皮素抑制病毒复制的机制可能与3C蛋白的活性有关[15]。
3.2调控细胞凋亡 病毒感染宿主后可导致宿主细胞发生凋亡。研究表明小RNA病毒的3C蛋白可通过激活半胱天冬酶(caspase)活性,裂解PinX1蛋白、真核翻译起始因子(eukaryotic translation initiation factor 4GI,eIF4GI)和受体相互作用蛋白激酶Ⅰ(Receptor-interacting protein kinase-1,RIPK1)调控细胞凋亡[16]。Li等研究结果显示,当神经细胞表达EV-71的3C蛋白时,细胞以DNA片段化和聚腺苷二磷酸核糖聚合酶(poly(ADP-ribose) polymerase,PARP)裂解的形式发生凋亡。其中,PARP裂解是caspase被激活的特异性标志,说明3C蛋白可激活caspase诱导细胞凋亡[17]。Song等发现EV-71感染宿主后,3C蛋白可与caspase -8,9相互作用而激活caspase-3诱发凋亡,当3C失去水解活性后,细胞caspase -8,9活性明显降低,细胞凋亡明显减少[18]。Chau等发现CV-B3的3C蛋白裂解宿主细胞的eIF4GI,导致细胞翻译过程受到抑制,最终细胞发生形态改变,皱缩等形式的凋亡[19]。Li等研究表明当EV-71感染宿主细胞时,3C蛋白能裂解PinX1蛋白中Q50-G51的特异性位 点,PinX1表达降低可使细胞DNA损伤,增加细胞凋亡的易感性[20]。与上述不同的是,Sarah等发现HRV-16的3C蛋白和caspase 8能裂解外在凋亡途径的关键中间体,即RIPK1。二者裂解RIPK1的位点和功能不同,caspase 8裂解RIPK1产生一个38 kD的蛋白,促进细胞发生早期凋亡,而3C蛋白进一步裂解38 kD的蛋白产生C末端23 kD的片段,抑制了凋亡的进一步发展[21]。
3.3逃避免疫应答 病毒感染宿主后,宿主启动固有免疫应答,如产生干扰素(interferon,IFN)和免疫因子抵御感染,而病毒通过一些策略逃避固有免疫应答的清除。研究表明3C蛋白通过抑制IFN的产生和核转录因子-κB(NF-κB)通路逃避免疫应答(图2)。Rui等研究发现CV-A6、CV-A16、和EV-D68等小RNA病毒感染细胞,3C蛋白能与黑色素瘤分化相关基因(Melanoma differentiation-associated gene 5,MDA5)、维甲酸诱导基因-I(Retinoic acid-inducible gene I,RIG-I)相互作用,破坏了RIG-I 和MAD5与通路下游线粒体抗病毒信号蛋白(mitochondrial antiviral signaling protein,MAVS)的结合,从而使MDA5、RIG-I介导的IFN I的产生受到抑制。3C蛋白还可抑制IRF3的磷酸化,从而抑制IFN I的产生[22]。Lei等研究表明EV-71的 3C蛋白与RIG-I的N端相互作用,抑制RIG-I与MAVS形成复合物,同时3C蛋白与β干扰素TIR结构域衔接蛋白 (TIR-domain-containing adaptor inducing interferon-β, TRIF)相互作用后诱导TRIF裂解,3C蛋白还可直接裂解IRF7 Q189和S190之间的位点,最终上述现象都抑制IFN I产生[23-25]。
NF-κB通路对于宿主抵抗病毒感染也至关重要。病毒感染后宿主可通过激活NF-κB通路,刺激机体产生抗病毒相关的免疫因子与细胞因子。3C蛋白可通过裂解TRAF家族与转化生长因子激酶1(transforming growth factor-βactivating kinase 1,TAK1)、TRIF、NF-κB相关激活因子(TRAF family member-associated NF-κB activator,TANK)抑制NF-κB通路活性。Lei等研究发现EV-71的3C蛋白裂解TAK1、TAK1结合蛋白1(TAK1 binding protein 1,TAB1)、TAB2和TAB3,破坏TAK1与TAB1、TAB2和TAB3形成复合物,从而抑制TAK 1复合物对NF-κB通路的激活和细胞因子的产生[26]。Xiang等研究表明在EV-D68中,作为半胱氨酸酶的3C蛋白可裂解TRIF的312和653位点,而导致TRIF失活。失活的TRIF抑制对NF-κB通路的激活,最终抑制宿主的免疫应答[27]。Huang等人研究EMCV发现3C蛋白能裂解TANK 蛋白的197和291谷氨酰胺位点。TANK即TRAF家族与NF-κB相关激活因子(TRAF family member-associated NF-κB activator,TANK),是NF-κB的关键调控因子。3C蛋白对TANK的裂解破坏这种抑制作用,可抑制TRAF-6介导的NF-κB通路的激活,这是一种新的小RNA病毒逃避宿主免疫的策略[28]。总而言之,小RNA病毒通过3C蛋白逃避免疫应答较为复杂,对3C蛋白的功能的探索有助于进一步阐明小RNA病毒的致病机制。
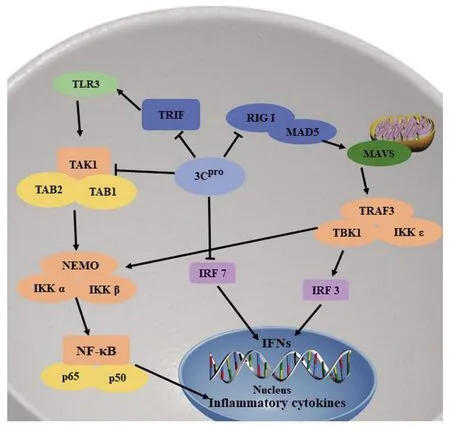
图2 小RNA 病毒3C蛋白调控免疫应答的机制Fig.2 Mechanisms of picornavirus 3C protein regulate the immune response
4 展 望
3C蛋白在小RNA病毒自身复制和与宿主的相互作用中发挥了重要作用。目前,3C蛋白抑制剂的研究是抗病毒药物的热点,研究小RNA病毒3C蛋白的功能,不仅能为深入研究小RNA病毒的致病机制奠定基础,还可为研发病毒疫苗以及抗病毒药物提供新的思路。
利益冲突:无
