高效液相色谱法测定淡水渔业用水中诺氟沙星、环丙沙星、恩诺沙星残留
2019-02-19孟丽华史艳伟时彦民简康刘方邓晨曦
孟丽华,史艳伟,时彦民,简康,刘方,邓晨曦
(济宁市渔业监测站,山东 济宁 272000)
诺氟沙星、环丙沙星和恩诺沙星3种喹诺酮类抗生素药物具有广谱、高效的特点,被广泛应用到水产养殖业中,起到了良好的效果[1-2]。但是调查发现在渔业养殖中使用的抗生素仅有小部分被鱼类吸收利用,还有大部分没有被食用或被食用后因没有被吸收以排泄物的方式排到水体中,在养殖水体中的大量残留,污染养殖环境的同时,对养殖渔业的健康发展也产生了不良影响[3-5]。因此,喹诺酮类抗生素药物的使用和监测应得到广泛的关注和高度的重视。但是,到目前为止对喹诺酮类药物残留的检测主要集中在水产品和海水养殖环境中,检测方法主要有液-质联用法[6-7]、高效液相色谱法[8-9]、酶联免疫法和荧光光度法等[10-11],国内外针对淡水渔业用水中的检测方法比较少见,对淡水渔业用水中的喹诺酮类抗生素药物的含量没有相关的限制值[12],也没有相关的国标、行标、地标,存在一定的监管盲区。
该次试验在参考相关文献的基础上,选取合适的富集和净化方法,对前处理条件和色谱条件进行优化,建立了用高效液相色谱-荧光法测定淡水渔业用水中的诺氟沙星、环丙沙星和恩诺沙星3种喹诺酮类抗生素药物,并用该方法对济宁地区的太白湖湿地、兖州兴隆庄和邹城西故煤炭塌陷地的渔业用水进行了检测。
1 材料与方法
1.1 淡水渔业用水采集
淡水渔业用水主要采集自济宁太白湖湿地、兖州兴隆庄煤炭塌陷地和邹城西故煤炭塌陷地等的养殖区,用采水器分别采集每个养殖水体的表层水500 mL,放在4℃冰箱内密封保存。
1.2 仪器设备与试剂
1.2.1 仪器设备 Agilent 1200型高效液相色谱仪,配荧光检测器;固相萃取装置(美国Agilent);talboys漩涡混合器(上海安普试验技术股份有限公司);KQ3200DE型数控超声波清洗器(昆山市超声仪器有限公司);Milli~Q超纯水器(上海默克化工技术有限公司);氮气吹干装置(美国Agilent);MP511型试验室PH计(上海三信仪表厂);Oasis HLB固相萃取小柱(3 mL/60 mg,Waters公司)。
1.2.2 标准品和试剂 诺氟沙星(纯度≥99.0%)、环丙沙星(纯度≥99.4%)、恩诺沙星(纯度≥99.7%)均购自TMtandard。甲醇、乙腈为色谱纯;三乙胺、磷酸为分析纯;试验过程中所用水均来自Milli~Q超纯水器。分别称取诺氟沙星标准品、环丙沙星标准品和恩诺沙星标准品各10.0 mg,用甲醇溶解并定容至100 mL的棕色容量瓶中,配置成浓度为100μg/mL的标准储备液,保存在-18℃冰箱中,保质期不超过6个月。
1.3 试验方法及步骤
1.3.1 样品前处理 量取经过0.45μm玻璃纤维滤膜过滤的水样50 mL,用稀盐酸调节pH值至4.0,然后用HLB柱进行富集、净化。将HLB分别依次用3 mL的甲醇和3 mL的纯水进行活化后,水样过柱(控制流速1 mL/min以内),先用3 mL的蒸馏水水淋洗,减压抽干后,用8 mL甲醇进行洗脱(控制流速1 mL/min),洗脱液在35~40℃下氮气吹干。吹干后用1.0mL的流动相进行定容,涡旋使得残渣充分溶解,过0.22μm水相滤膜,待测。
1.3.2 色谱方法 色谱柱:Water X BridgeC18柱(250 mm×4.6 mm,5μm);流动相:A为500 mL水加入3 mL磷酸混和均匀后用三乙胺调节pH值至2.4,B 为乙腈(A:B=82:18);流速:1.0 mL/min;柱温:40℃;进样量20μL;检测器:荧光检测器;激发波长为280 nm;发射波长为450 nm。
1.3.3 标准曲线的制备 准确量取一定量的标准储备液,用流动相将诺氟沙星、恩诺沙星稀释成浓度分别为2.5、5.0、10.0、25.0、50.0、100.0、250.0 μg/L的标准工作液、将环丙沙星稀释成浓度为0.5、1.0、2.0、5.0、10.0、20.0、50.0 μg/L 的标准工作液待用。按浓度由低到高依次进样,每一个浓度重复进样2次取平均值,以峰面积的平均值为纵坐标,标准溶液的浓度为横坐标,绘制标准曲线,计算回归方程和相关系数。
1.3.4 检出限和定量限的计算 在不含3种喹诺酮类抗生素待测组分的空白渔业用水中加入诺氟沙星、环丙沙星、恩诺沙星标准储备液,处理分析后,以浓度为横坐标,峰面积为纵坐标绘制标准曲线,计算3倍信噪比时对应的浓度为分析物的最低检测浓度,即方法的检出限;10倍信噪比时对应浓度为分析物的最低定量浓度,即定量限。
1.3.5 回收率和精密度的计算 选用不含3种喹诺酮类抗生素待测组分的渔业用水空白样品,制成3组不同浓度的标准添加样品,其中,诺氟沙星、恩诺沙星在5.0、25.0、50.0μg/L 3个浓度水平进行加标回收试验,环丙沙星在1.0、5.0、10.0μg/L 3个浓度水平进行加标回收试验,每个添加浓度进行6个平行试验,按照“1.3.1”和“1.3.2”的方法步骤进行处理和分析,计算回收率和相对标准偏差。
2 结果与讨论
2.1 前处理条件的优化
在参考相关文献的基础上,该文对固相萃取柱、上样pH值及体积、洗脱试剂及其体积等对试验结果有影响的因素进行进行了分析讨论,确定了前处理的最优条件。
2.1.1 固相萃取柱的选择 该文分别采用药残净化和富集最常用的固相萃取柱C18小柱和HLB小柱进行了试验,以净化效果和回收率作为评价依据。在其他条件不变的前提下,用不同的萃取柱进行试验,结果表明,采用HLB小柱萃取的样品回收率略高于C18小柱,且图谱干净,杂峰较少,因此,该方法采用HLB小柱对样品净化。
2.1.2 上样pH值 由于固相萃取的效率会受到样品pH值的影响,因此该文对上样pH进行了优化。一般淡水的pH值在6.5~8.5,以空白的淡水渔业用水为基质,调节pH值,使得pH值分为3.0、4.0、5.0、6.0、7.0、8.0、9.0 然后分别加入目标喹诺酮类抗生素混合标准溶液,混合均匀后采用“1.3.1”的提取方法和步骤进行处理,在不考虑其他因素的干扰的情况下以加标回收率为评价依据最终发现当pH为4.0时3种喹诺酮类抗生素的回收率较高,并且重现性较好。
2.1.3 上样体积 以过滤后的淡水渔业用水为基底,加入3种喹诺酮类抗生素混合标准溶液,分别考察 10、20、30、40、50、60、70、80 mL 上样体积对回收率的影响。3种喹诺酮类抗生素都在上样体积为50 mL时回收率达到最大值,之后有趋于平稳,因此最终上样体积选择50 mL。
2.1.4 洗脱试剂及其体积 为了降低基质效应,试验对乙腈和甲醇的洗脱效果进行了比较。当用乙腈做洗脱剂时,加标样品中的诺氟沙星回收率为48.2%、环丙沙星回收率为53.7%、恩诺沙星的回收率为52.5%;当用甲醇做洗脱剂时加标样品中的诺氟沙星的回收率为75.5%、环丙沙星的回收率为78.2%、恩诺沙星的回收率为73.5%。由此看见用甲醇做洗脱剂时回收率较高,所以该试验选择的洗脱剂为甲醇,根据结果比较,甲醇洗脱体积定为8 mL。
2.2 色谱条件的优化
因为喹诺酮类化合物同时具有酸性基团和碱性基团,易形成拖尾,从而使得诺氟沙星和环丙沙星很难在短时间内完全分离,而加入三乙胺作为改良剂能够改善拖尾现象。在用三乙胺调节流动相的pH值时,发现pH值在2.4时3种目标物可以在合适的时间分离完全。同时,流动相中的乙腈含量对保留时间影响很大,经过对乙腈和磷酸盐不同比列的试验,发现当乙腈:磷酸盐=18:82时诺氟沙星、环丙沙星、恩诺沙星的出峰时间在6~10 min,而样品中的杂峰会集中出现在6 min之前,不会对目标物造成干扰。
2.3 标准曲线和检出限
按1.3.3方法和步骤绘制标准曲线,按1.3.4方法和步骤计算检出限,线性方程、相关系数、检出限和定量限列于表1。
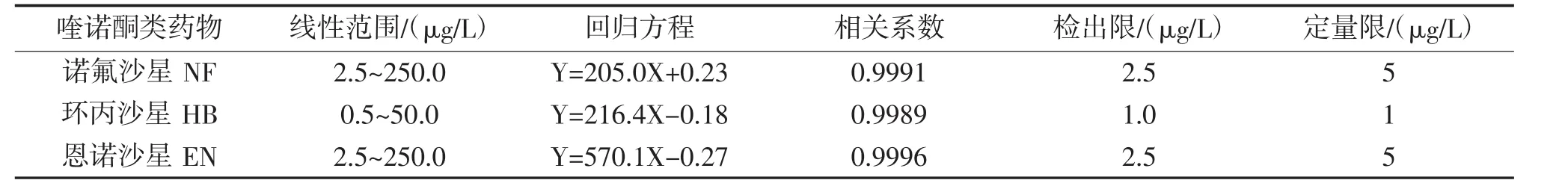
表1 3种喹诺酮类药物线性范围和检出限
2.4 回收率与精密度
按1.3.5方法和步骤计算回收率和精密度,结果见表2,其中诺氟沙星的回收率为85.9%~92.6%,相对标准偏差为3.77%~6.73%;环丙沙星的回收率为78.0%~84.4%,相对标准偏差为3.82%~6.26%;恩诺沙星的回收率为81.0%~89.9%,相对标准偏差为3.65%~7.73%。
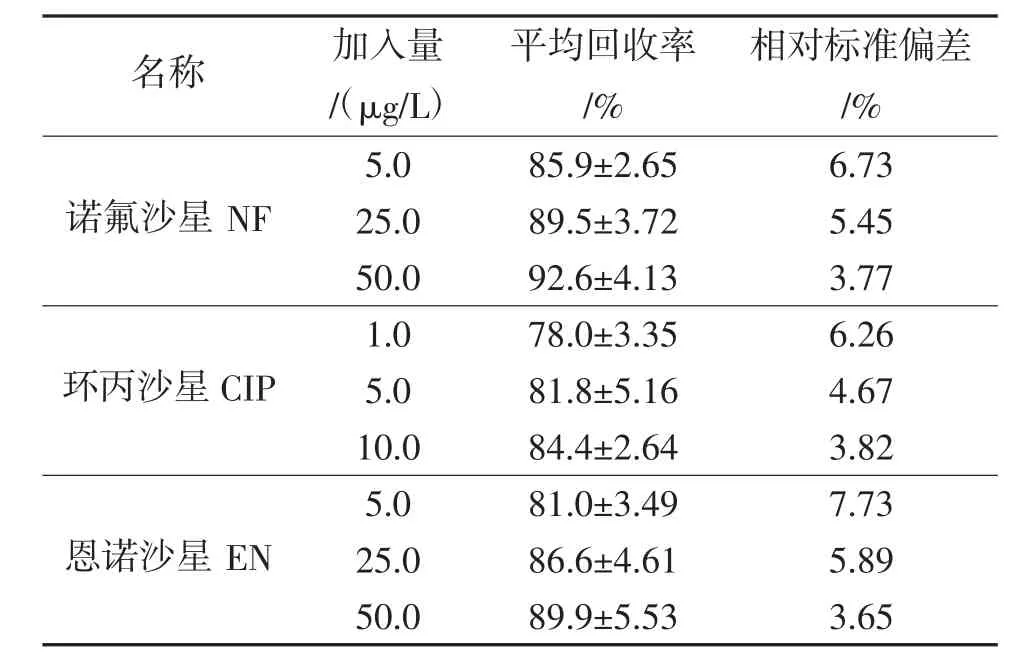
表2 加标回收率和相对标准偏差
2.5 液相色谱图
空白淡水渔业用水、淡水渔业用水加标样品(诺氟沙星、恩诺沙星12.5μg/L,环丙沙星2.5μg/L)、标准样品(诺氟沙星、恩诺沙星12.5μg/L,环丙沙星2.5μg/L)的色谱图分别见图1中的a、b、c。由图可见,在1.3.2色谱条件下,3种抗生素分离效果很好。
2.6 实际样品的测定
从济宁太白湖湿地、兖州兴隆塔煤炭塌陷地、邹城西固煤炭塌陷地等3个地区的9个养殖场采集渔业用水9份,按1.3.1前处理步骤和1.3.2的检测方法对采集的水样进行处理和检测分析,9个养殖场的渔业用水中3种喹诺酮类药物均低于检出限。
3 结论
该研究的结果表明高效液相色谱-荧光法适用于淡水渔业用水中喹诺酮类抗生素含量的检测,此方法专属性强、灵敏度高,净化和分离效果好,相对于液-质联用仪的检测方法具有操作简单,成本低的优点。此法可应用于淡水养殖地区不同渔业用水中的喹诺酮类抗生素含量的测定,为养殖用水中喹诺酮类抗生素药物的生态风险评估提供科学的依据。
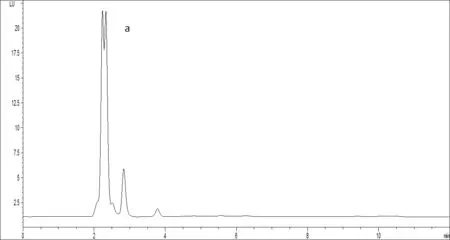
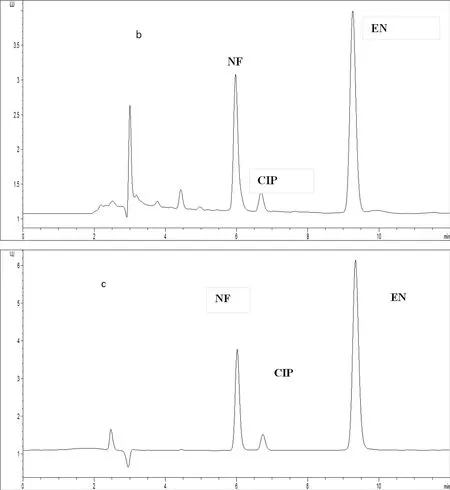
图1 喹诺酮类抗生素的液相色谱图
