天然橡胶热解产物反应机理研究
2019-02-14杨启容邹瀚森魏鑫姚尔人钟浩文禹国军
杨启容,邹瀚森,魏鑫,姚尔人,钟浩文,禹国军
(1.青岛大学机电工程学院,266071,山东青岛;2.西安交通大学能源与动力工程学院,710049,西安;3.上海交通大学船舶海洋与建筑工程学院,200240,上海;4.上海海事大学商船学院,201306,上海)
目前,废旧橡胶制品(如废旧轮胎、废橡胶手套、废胶带等)在废旧高分子材料中是仅次于废旧塑料的一大类固体废弃物,其中废旧轮胎占废旧橡胶的70%左右[1],而废旧轮胎的主要成分是天然橡胶、合成橡胶、炭黑以及多种助剂[2],由于轮胎中的橡胶不易被化学物质和细菌腐蚀和分解,需要超过一百年的时间才能销毁[3],这导致了大量废旧轮胎被搁置,不仅造成了大量土地的占用,而且还容易引发火灾[4-5]。然而,轮胎中的橡胶具有高于煤炭的热值,大约为29~37 MJ/kg[6],所以合理利用废旧轮胎不仅能够保护环境,并且能实现资源的再利用。
在现有的轮胎利用方式中,热解作为一种比较有效的回收手段,不仅可以将难以处理的废旧物品分解,而且能够生产可重复利用的产品[7],其热解的产品有热解气(C1~C4的碳氢化合物、H2等)、热解油(C5以上的碳氢化合物)以及固体产物(炭黑等)[8],其中气体产物具有较高的热值,可为热解装置提供热量和为其他工厂供能[1],液体产物可以作为燃料油及通过分离提取轻质、中质和重质油,形成的固体炭可以用作活性炭和炭黑[9-11]。
目前,部分学者通过实验手段对橡胶热解的过程、产物、动力学及改变条件对热解产物的影响规律开展了研究,Kordoghli等用MgO、Al2O3、CaCO3、分子筛ZSM-5分别与牡蛎壳混合后来催化废轮胎热解,发现产生的气体量比传统热解高45%,热值高16%[12];Abnisa等用棕榈壳与轮胎共热解,发现可以增加液体和气体的产量,减少固体的产量,当棕榈壳和轮胎混合添加比为1∶1时最佳[13]。但是,实验手段无法分析产物形成过程中复杂的反应过程,而计算机模拟手段可以解决这个问题,其中反应分子动力学已经被广泛应用到聚合物热解机理的研究中。Liu等基于反应动力学和密度泛函理论研究了甲基环己烷高温热解下的反应机理,求得的相应反应活化能与实验结果一致[14];Zhao等通过反应分子动力学探究了聚碳酸酯热老化的反应机理,通过模拟计算得到的活化能及老化的反应机制与实验结果吻合[15]。因此,可以用模拟手段来对天然橡胶热解的气相产物的反应机理进行研究。
通过实验已经证明了在天然橡胶初级热解(300~550 ℃)的过程中,主要产物有异戊二烯单体和二聚体-1,4-二甲基-4-乙烯基环己烯[16-17],但是对其进一步热解生成小分子气体的热解过程并不了解。为了更好地探究其反应机理,本文在单体和二聚体模型的基础上,采用密度泛函理论对气体产物可能生成的路径进行模拟,并对每条路径的反应物、中间体和生成物进行热力学和动力学计算,再进一步计算得到各自由基反应的速率常数。本文采用Aspen plus建立一维裂解反应器,以所得到的动力学数据为模型进行反应过程模拟,得到原料和产物沿管长的浓度变化,通过对结果进一步分析得到其反应机理,最终验证了气体的反应路径。
1 研究方法
1.1 MS软件模拟计算
首先在Visualizer模块中构建单体和二聚体等反应物、中间体(包括各自由基团)和产物(气体小分子)模型,然后在DMoL3模块中通过哈密尔顿函数GGA/BP[18]对反应物、中间体和各种产物模型进行几何优化,并对模型进行振动频率计算,若没有出现虚频,表明此时得到的结构在势能面上具有极小值。通过振动分析后的结果可以用于计算焓、熵、自由能和等压热容等分子系统的热力学参数,并经过热力学分析可以得到零点总电子能和离子能Etot及内能U,各热力学量之间关系如下。
系统的内能来源于电子的振动、平移和旋转,计算式为
U=Evib+Etrans+Erot
(1)
式中:Evib、Etrans、Erot分别表示振动、平移和转动对能量的贡献。焓H的计算式为
H=Etot+U
(2)
吉布斯自由能G的计算式为
G=H-TS
(3)
式中:T表示温度;S表示熵。若反应式为

(4)
则该反应的焓变ΔH为
ΔH=HB+HC-HA
(5)
式中:HA、HB、HC分别表示物质A、B、C的焓值。
采用DMol3中的LST/QST(线性同步变换/二次同步变换)工具搜索反应物与产物之间的过渡态,并进行频率计算,如果在红外谱上只出现一个虚频,表明得到的是过渡态[19],然后对过渡态进行几何优化,得出该反应的反应势垒,进行相应的动力学分析。通过得到的数据,由下式[19]计算各自由基反应的反应速率常数,即
(6)

1.2 裂解工艺过程模拟
采用Aspen plus V8.4软件模拟单体的裂解工艺过程,选用Tempt运行环境,采用RPlug反应器中的ICON2模型构建一维反应器模型[19-20]。首先在Properties模块中对自由基物性进行分析,然后将所有反应及其反应动力学数据(包括反应势垒和反应速率常数)输入到reaction模块中,并在stream模块中设定进料条件,在block模块中设定反应器的规格、壁温及压力(参数条件见表1),通过调节入口温度,可使原料在反应器中基本反应完成,根据获得的各自由基浓度变化情况对产物分布等数据进行分析,确定主要的热解路径。

表1 工艺过程模拟的操作条件
2 结果与讨论
天然橡胶的热解实验表明,气体产物有甲烷、乙烷、乙烯、丙烷、丙烯、丁二烯、苯、甲苯、二甲苯、苯乙烯、氢气等[16],其中氢气和甲烷在气体中所占的比重较大[9],并且随热解温度的升高,氢气的产量会进一步增大。
2.1 单体热解过程
在热解过程中,主链上连接两个基元的C—C键首先发生断裂,生成C5H8基,然后首尾两端的C—C单键缩合成双键形成C5H8单体。图1是异戊二烯单体各键长的数据,可以看出C3—C5的键长比其他碳碳之间的键长要长,C5—H、C2—H之间的键长比其他碳氢之间的键长要长,所以可以推测异戊二烯链上C3—C5、C5—H、C2—H键更容易断裂,并结合相关文献[21-24]推断出单体可能发生的5条反应路径。
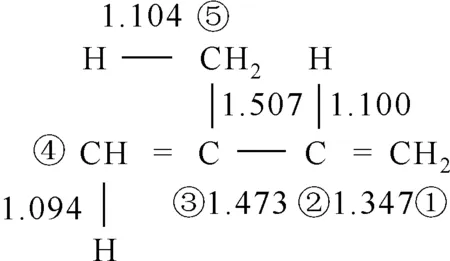
图1 异戊二烯单体键长数据
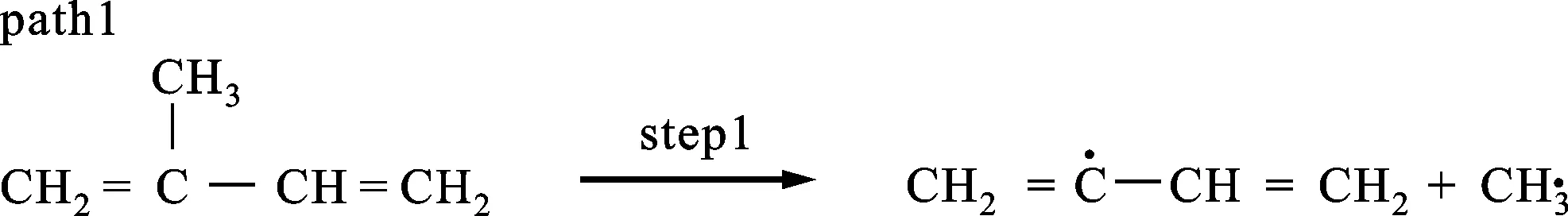
(a)链式引发反应

(b)氢自由基攻击单体夺取氢
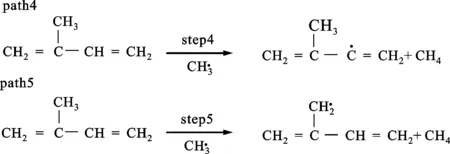
(c)甲基攻击单体夺取氢图2 异戊二烯单体(C5H8)的热解反应路径
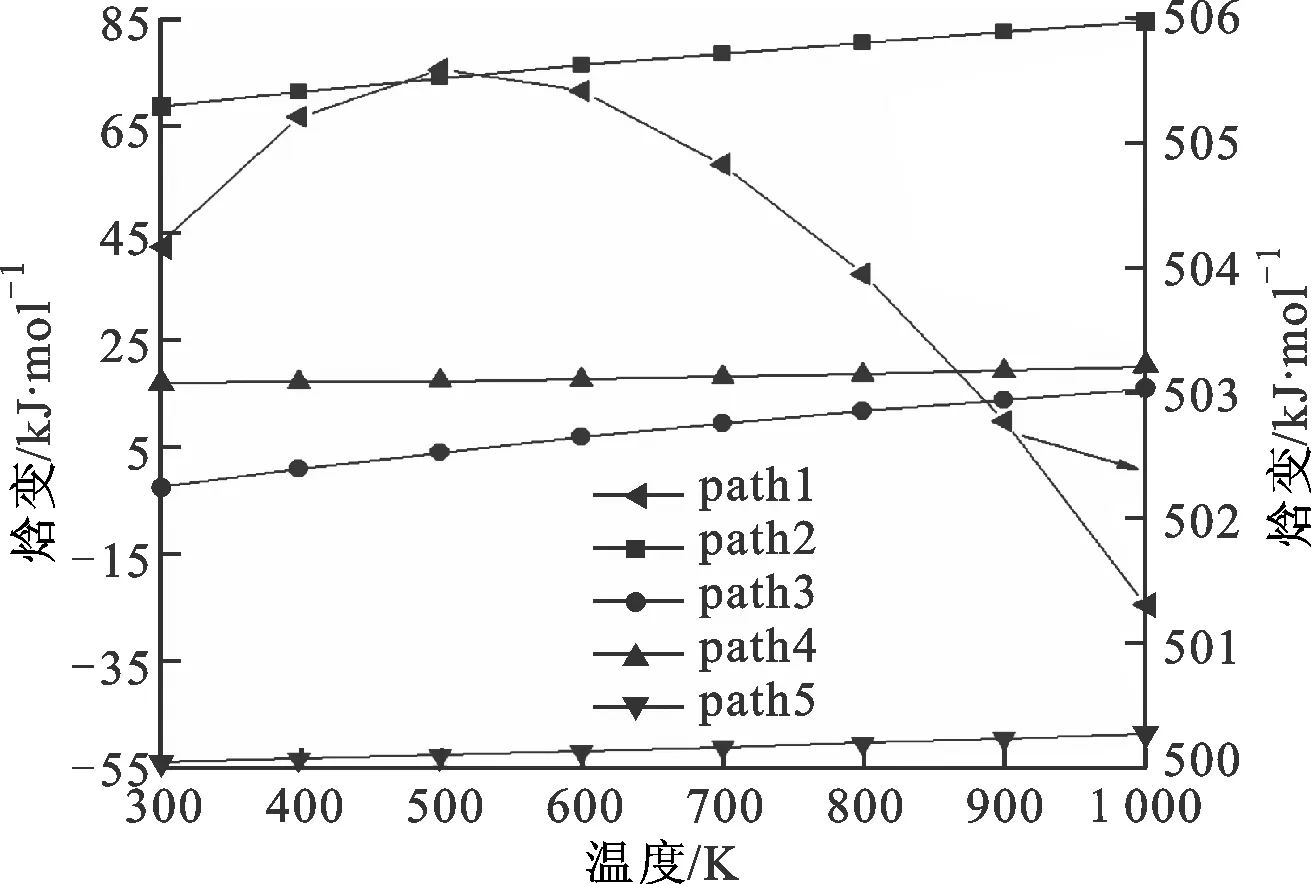
图3 单体各反应路径的焓变随温度的变化

图4 单体各反应路径的吉布斯自由能变随温度的变化
图2给出了单体的5条热解反应路径,图3是每条路径的焓变ΔHk随温度的变化情况,图4是自由能变ΔGk随温度的变化情况。表2是通过GGA/BP算法对图2中的5条路径的各反应物、产物、过渡态经过几何优化后得到的能量及反应势垒。图2a是path1的热解过程,单体上的C3—C5键发生断裂,生成了活性较强的甲基和比较稳定的丁二烯基,这一步的反应势垒为437.50 kJ/mol,与文献中的375.06 kJ/mol相比误差为16.5%(见表2),而且从图3中可以看出path1的ΔHk基本在500 kJ/mol以上,远大于其他4条路径,并且温度为500 K时出现了最大值。经过分析,发现相对质量小的分子,温度低时对其焓值影响程度较小,变化范围不大,温度高时变化范围加大,此路径中有甲基,在对产物和反应物作差时就出现了一个最大值。从图4中也可以看出,path1的ΔGk都位于320 kJ/mol以上,远高于其他4条路径。图2b中path2、3都是发生的氢自由基攻击单体夺取氢的反应,不过path2是活性较强的氢自由基攻击单体C5H8,导致乙烯基上的C—H键发生断裂,然后与氢自由基结合生成H2,此步的反应势垒为21.37 kJ/mol,而path3则是氢自由基攻击单体C5H8,导致丙烯基上的烯丙基键C—H键发生断裂,然后与氢自由基结合生成H2,此步的反应势垒为5.78 kJ/mol。通过比较path2与3可以看出,path3的势垒小于path2的势垒,说明破坏烯丙基键比破坏乙烯基键更容易。这是因为乙烯基键C—H的键能(412 kJ/mol)大于烯丙基键C—H的键能(362.5 kJ/mol)。从图3、4中也可以看出,path3中在每个温度下的ΔHk和ΔGk都小于path2,这都说明path3比path2更有竞争力。图2c中的path4、5发生的则是甲基攻击单体夺取氢的反应,其中path4是甲基攻击单体C5H8,导致乙烯基上的C—H键发生断裂,然后与氢自由基结合生成CH4,这一步的反应势垒为50.07 kJ/mol,与文献中的46.67 kJ/mol比较接近(见表2);path5则是甲基攻击单体C5H8,导致丙烯基上的烯丙基键C—H键发生断裂,然后与氢自由基结合生成CH4,这一步的反应势垒为18.45 kJ/mol,接近于文献中的19.18 kJ/mol(见表2)。比较发现,path5的势垒小于path4的势垒,说明path5比path4有竞争力,也进一步证明了乙烯基键比烯丙基键稳定。图3、4中的ΔHk和ΔGk也是path5的值都要小于path4的值。比较path3、5可以发现,同样是与丙烯基上脱离的氢自由基结合,path3需要突破的势垒小于path5的势垒,说明氢自由基的活性强于甲基的活性。通过比较5条反应路径的势垒发现,path1>path4>path2>path5>path3,所以从动力学角度看,优先支持的路径是path3。同样地,按ΔGk从大到小的顺序排列:当温度低于850 K时,path1>path4>path2>path5>path3,热力学上优先支持的路径顺序为path3>path5>path2>path4>path1;温度高于850 K时,path2要优于path5。结合动力学和热力学计算结果可以看出,在单体的热解过程中,path3是主要的热解反应通道,H2是主要的产物。
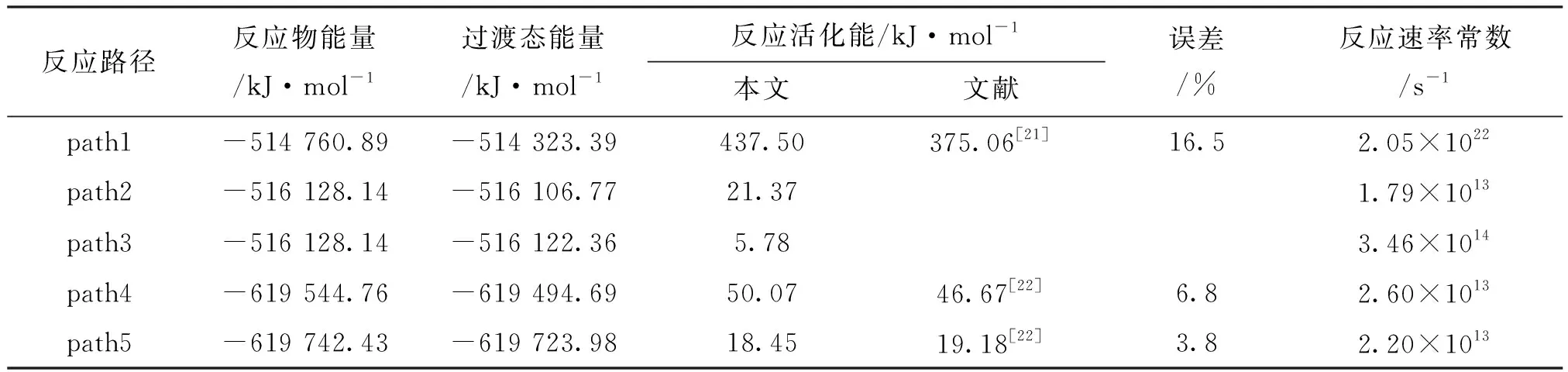
表2 单体各反应物、过渡态经过零点能矫正后的总能量、反应活化能及对比误差
注:反应活化能=过渡态能量-反应物能量。
从图2中也可看出,除了生成H2和CH4外,还有甲基、C4H5基、C5H7基产生,这些基团会进一步相互结合生成其他分子,也可以再次生成单体,反应式如下

(7)
(8)
(9)
(10)
(11)
2.2 二聚体热解过程
初始热解过程中生成的二烯烃受热会发生重排,进一步通过环化反应生成1,4-二甲基-4-乙烯基环己烯等环烯烃。随着温度继续升高,二聚体等大分子体系会继续热解,生成含C较小的分子,所以以二聚体为模型,结合相关文献[25]推测出二聚体可按图5中的4条路径进一步热解。
图5、6分别是二聚体的反应路径及其焓变随温度变化的情况。path6、7都是二聚体首先发生开环反应生成两个C5H8基(即M1和M2),然后path6发生的是M1两端的碳碳单键缩合成双键形成异戊二烯单体,这步的反应势垒为373.04 kJ/mol。在图6中可以看到ΔHk都是正值,说明是一个吸热反应。path7则是在碳碳单键缩合成双键之前受到甲基的攻击,被夺取一个氢生成了C5H7基和CH4,该步的反应势垒是354.48 kJ/mol。从图6中可以看到ΔHk都是负值,说明是一个放热反应。path8是二聚体经过step4,C—C单键发生了断裂,生成二甲基环已烯基M3和乙烯基M4,然后M3的C—C单键缩短成CC双键,形成了二甲苯,该步的反应势垒为985.11 kJ/mol。path9是一个甲基从六元环上脱离出来,然后形成的甲基-乙烯基环已烯基M5进一步分裂成丁二烯和异戊二烯,该步的反应势垒为649.95 kJ/mol。从图6中可以发现path8、9的ΔHk比较接近,都大于500 kJ/mol,高于另外两条路径,说明path8、9反应比较困难,并且两条路径的数值都存在最大值。分析发现相对质量小的分子,温度低时对其焓值影响程度较小,变化范围不大,温度高时变化范围加大,这2条路径的产物中都包含小分子体系甲基和乙烯基,在对产物和反应物作差时就出现了一个最大值。比较4条路径的势垒,发现在动力学上优先支持的是path7。图7给出了相应的每条路径的ΔGk随温度的变化,可以看出,按ΔGk从大到小的排序为path8>path9>path6>path7,所以热力学优先支持的路径顺序为path7>path6>path9>path8。综合动力学和热力学计算,path7是二聚体热解的主要通道。
而且都远远
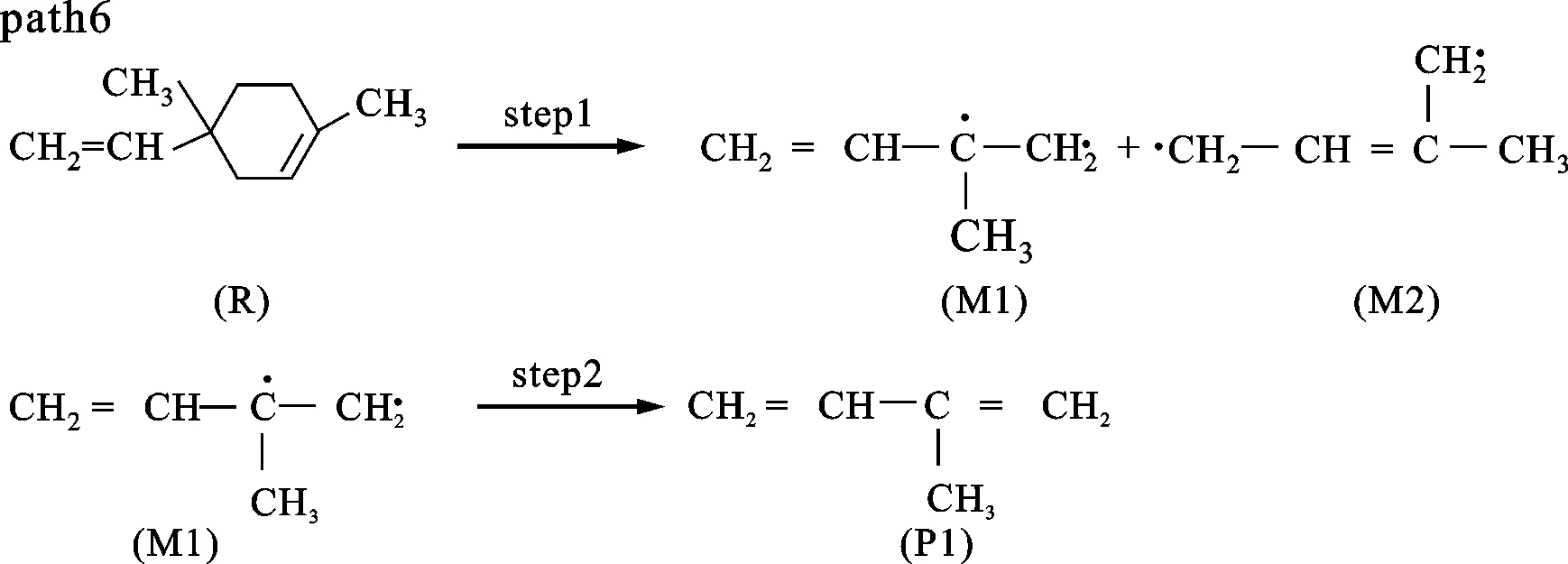
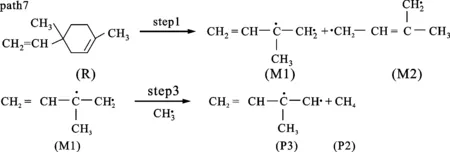
(a)单体、CH4生成的可能路径

(b)二甲苯生成的可能路径
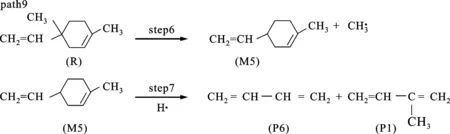
(c)单体及丁二烯生成的可能路径图5 二聚体C10H16后续的热解反应路径
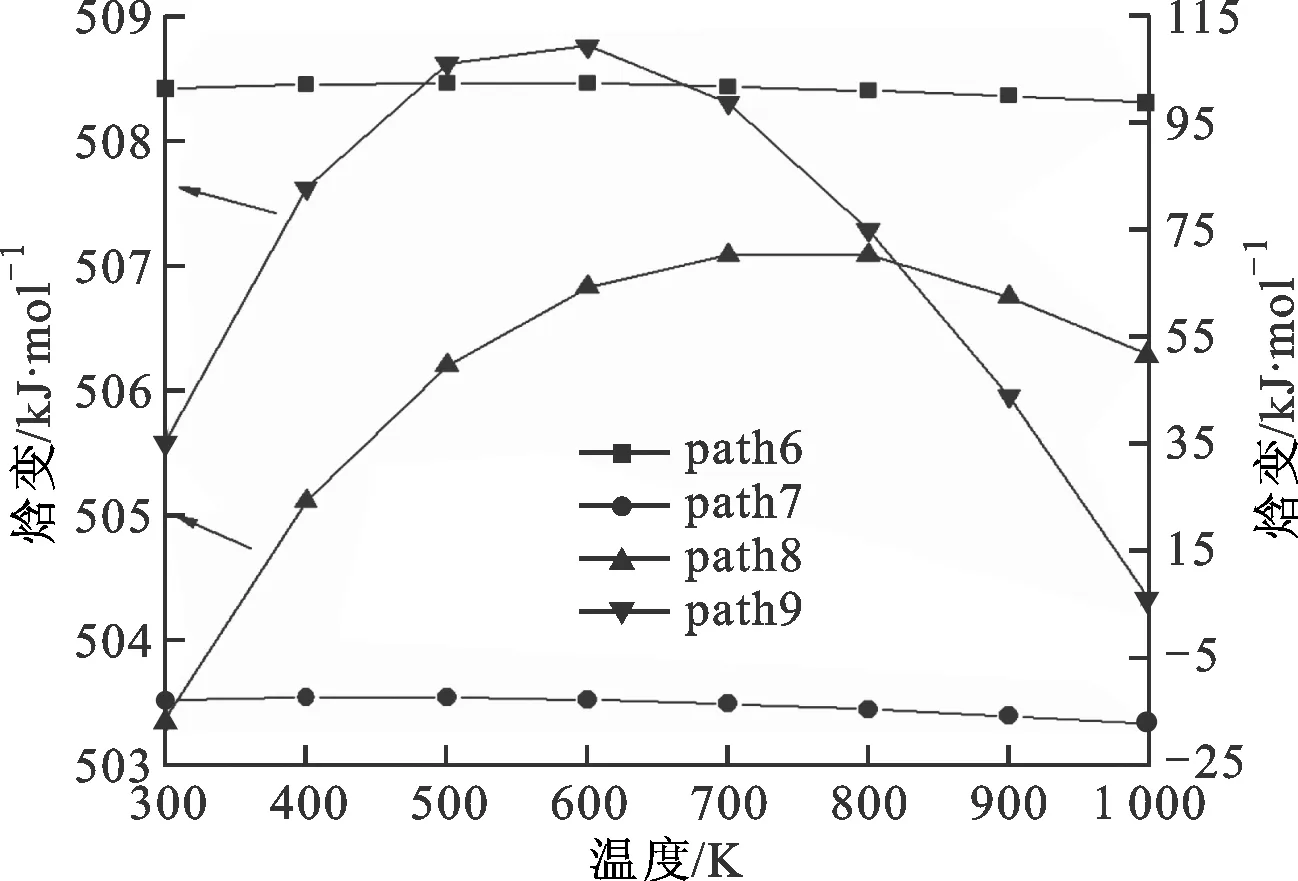
图6 二聚体各反应路径的焓变随温度的变化
在热解过程中还存在着大量的乙烯基、甲基、氢自由基等,它们也会相互结合生成其他的小分子气体,如下式所示
(12)
(13)
(14)

图7 二聚体各反应路径的吉布斯自由能变随温度的变化
2.3 热解工艺过程模拟

图8 总反应中各原料及产物的体积分数变化情况
采用Aspen plus对单体C5H8热解反应(path1~5)进行模拟计算,温度为750 K时的结果如图8所示。从图中可以看出,原料有单体C5H8、氢自由基和甲基,对应图2中的反应物。在单体热解过程中,path1发生的是单体断裂生成甲基和丁二烯基。从图8中可以看到:C4H5含量非常小,曲线基本与横坐标重合,说明path1发生的可能性很小;path2、3、4、5都是自由基与单体发生取代的反应,其中path2、3是氢基作为自由基,而path4、5则是甲基为自由基,对应的产物分别是氢气及异戊二烯基和甲烷及异戊二烯基,通过比较氢自由基和甲基含量的变化,发现氢自由基随反应进行变得越来越少,直至完全反应,而甲基曲线变化幅度很小,并且当氢自由基反应完后,单体含量也基本不再变化,说明氢自由基比甲基更容易与单体发生取代反应,即path2、3比path4、5更容易发生;对于path2、3,虽然产物成分相同,但是单体失去氢的位置不同,为了进一步作比较,将path2、3生成的C5H7分别记为C5H7-1和C5H7-2,通过比较曲线得到C5H7-2的生成量大于C5H7-1,大约是其的2.5倍,说明path3比path2更有可能发生。模拟结果与MS的计算结果吻合。
3 结 论
(1)在单体的5条路径中,path1、2、3、4、5的活化能分别为437.50、21.37、5.78、50.07、18.45 kJ/mol,比较得出path3即由氢自由基夺取单体C5上的H生成H2,无论是热力学参数还是动力学参数都是相对较小的,是单体后续热解过程中发生可能性最大的路径,相应的产物H2在气体成分中所占的比重应该也是较大的。
(2)二聚体的4条路径活化能分别是373.04、354.48、985.11、649.95 kJ/mol,比较得出二聚体中由二聚体先发生开环反应生成两个C5H8基,然后被甲基夺取氢进一步生成CH4的path7,是二聚体后续热解过程中发生可能性最大的路径,相应的产物CH4在气体成分中所占的比重应该也是较大的。
(3)通过Aspen plus软件对单体的所有路径进行模拟计算,根据产物的分布情况得出H2的含量最高,其中path3产生的H2含量大约是path2的2.5倍。裂解反应模型分析的结果与动力学和热力学得出的结果是吻合的,证明推导的反应路径是合理的,这为废旧轮胎的气化回收利用提供了理论支持。
