Deferoxamine promotes recovery of traumatic spinal cord injury by inhibiting ferroptosis
2019-02-13XueYaoYanZhangJianHaoHuiQuanDuanChenXiZhaoChaoSunBoLiBaoYouFanXuWangWenXiangLiXuanHaoFuYongHuChangLiuXiaoHongKongShiQingFeng
Xue Yao , Yan Zhang , Jian Hao , Hui-Quan Duan , Chen-Xi Zhao , Chao Sun , Bo Li , Bao-You Fan , Xu Wang , Wen-Xiang Li , Xuan-Hao Fu , Yong Hu, Chang Liu, Xiao-Hong Kong, Shi-Qing Feng ,
1 Department of Orthopedics, Tianjin Medical University General Hospital, Tianjin, China
2 State Key Laboratory of Medicinal Chemical Biology, Nankai University, Tianjin, China
3 International Science and Technology Cooperation Base of Spinal Cord Injury, Tianjin, China
4 Department of Orthopedics, Nankai Hospital, Tianjin, China
5 Department of Orthopedic and Traumatology, The University of Hong Kong, Pokfulam, Hong Kong Special Administrative Region, China
6 School of Medicine, Nankai University, Tianjin, China
7 Tianjin Neurological Institute, Key Laboratory of Post-Neuroinjury Neuro-repair and Regeneration in Central Nervous System, Ministry of Education and Tianjin City, Tianjin, China
Abstract Ferroptosis is an iron-dependent novel cell death pathway. Deferoxamine, a ferroptosis inhibitor, has been reported to promote spinal cord injury repair. It has yet to be clarified whether ferroptosis inhibition represents the mechanism of action of Deferoxamine on spinal cord injury recovery. A rat model of Deferoxamine at thoracic 10 segment was established using a modified Allen's method. Ninety 8-week-old female Wistar rats were used. Rats in the Deferoxamine group were intraperitoneally injected with 100 mg/kg Deferoxamine 30 minutes before injury. Simultaneously, the Sham and Deferoxamine groups served as controls. Drug administration was conducted for 7 consecutive days. The results were as follows: (1) Electron microscopy revealed shrunken mitochondria in the spinal cord injury group.(2) The Basso, Beattie and Bresnahan locomotor rating score showed that recovery of the hindlimb was remarkably better in the Deferoxamine group than in the spinal cord injury group. (3) The iron concentration was lower in the Deferoxamine group than in the spinal cord injury group after injury. (4) Western blot assay revealed that, compared with the spinal cord injury group, GPX4, xCT, and glutathione expression was markedly increased in the Deferoxamine group. (5) Real-time polymerase chain reaction revealed that, compared with the Deferoxamine group, mRNA levels of ferroptosis-related genes Acyl-CoA synthetase family member 2 (ACSF2) and iron-responsive element-binding protein 2 (IREB2) were up-regulated in the Deferoxamine group. (6) Deferoxamine increased survival of neurons and inhibited gliosis. These findings confirm that Deferoxamine can repair spinal cord injury by inhibiting ferroptosis. Targeting ferroptosis is therefore a promising therapeutic approach for spinal cord injury.
Key Words: nerve regeneration; iron; spinal cord injury; secondary injury; ferroptosis; deferoxamine; GPX4; xCT; treatment; astrogliosis; lipid peroxidation; neural regeneration
Introduction
There is currently no effective cure for acute traumatic spinal cord injury (SCI), and our understanding of the molecular mechanisms of SCI is limited (Wu et al., 2012; Ropper and Ropper, 2017; Fan et al., 2018). Many studies have investigated the mechanism of cell death and have developed novel therapies in SCI. Although many cell death pathways, such as apoptosis, necroptosis, and autophagy, have been identified (Wang et al., 2014b; Fan et al., 2016; He et al., 2016), the major cell death pathway contributing to the imbalanced microenvironment in SCI is still unknown. Cell death of motor neurons has been found to occur with loss at the epicenter by 4 hours in rats (Grossman et al., 2001a, b). For oligodendrocytes, half of them died within the first 4 hours post injury,with little subsequent loss (Liu et al., 1997). Apoptosis is an inflammatory-limited cell death pathway and therefore may not contribute much to the inflammatory microenvironment,which hinders regeneration (Lu et al., 2000). Therefore, our current understanding of cell death in SCI cannot explain its pathophysiology. There may be other forms of cell death that contribute to SCI-induced secondary injury. Ferroptosis, a newly identified iron-dependent cell death pathway, shares several features with the pathogenic changes of SCI. First,iron overload triggers ferroptosis (Dixon et al., 2012). The acute phase of traumatic SCI involves immediate hemorrhage and reactive oxygen species accumulation (Choi et al., 2012;Xiao et al., 2015). Iron is overloaded at the injury site due to hemorrhage (Hao et al., 2017). Second, ferroptosis is also initiated by lipid reactive oxygen species (Imai et al., 2017).As the spinal cord contains a high content of polyunsaturated fatty acids, its lipid peroxidation contributes to pathologies of SCI (Bazinet and Laye, 2014). Third, glutamate-induced toxicity which can also induce ferroptotic cell death in vitro in brain slices plays a key role in the pathophysiology of SCI(Yu et al., 2009; Dixon et al., 2012). Given that iron-overload,lipid reactive oxygen species, and glutamate accumulation are all present in SCI, we hypothesized that the ferroptosis pathway plays an important role in secondary injury of SCI.
Deferoxamine (DFO), a potent iron chelator, is approved by Food and Drug Administration and used for treating diseases with iron overload. While the neuroprotective effect of DFO has been confirmed, its underlying mechanism remains elusive. The relationship between iron chelation and reduced cell death is not understood. Recently, DFO was identified as a ferroptosis inhibitor (Dixon et al., 2012; Xiao et al., 2015). Therefore, we explored whether DFO promotes SCI repair through ferroptosis inhibition and aimed to elucidate the role of ferroptosis in the secondary pathophysiology of SCI.
Materials and Methods
Animals
Ninety female Wistar rats aged 8 weeks and weighing 240± 10 g were obtained from Experimental Animal Center of the Academy of Military Medical Sciences, Beijing, China[license No. SCXK (Jin) 2014-0002]. Rats were kept under a humidity- and temperature-controlled environment with a 12-hour light/dark cycle, and had free access to food and water. All experimental procedures on animals were approved by the Ethics Committee of Institute of Radiation Medicine Chinese Academy of Medical Sciences (Tianjin, China) (approval number: DWLI-20171010) in accordance with the guidelines provided by the Committee for Purpose of Control and Supervision of Experiments on Animals (CPCSEA).
Experimental design
This study contained three study arms with different survival times following SCI. Rats were randomly assigned to three groups, as follows: the Sham group (n = 30), which received laminectomy only without SCI; the SCI group (n = 30), which received T10 contusion injury with saline treatment via intraperitoneal injection; and the DFO group (n = 30), which received T10 contusion injury with DFO 100 mg/kg treatment via intraperitoneal injection, modified from previous report(Hao et al., 2017). The rats were sacrificed at 1, 4, 8, 24 hours, 2,3, 7 days, 2 or 8 weeks after SCI. After transcranial perfusion,spinal cord tissues of rats were collected and processed for molecular and biochemical analysis (Figure 1A).
Contusion SCI models
A contusion SCI model was established using a modified Allen's method (Koozekanani et al., 1976). The rats were deeply anesthetized with 3 mL/kg 4% chloral hydrate by intraperitoneal injection. An approximately 1-cm midline incision along dorsal skin was made and the muscle layers over the area of the vertebral T10 level were bluntly dissected to expose T10 vertebral laminae. Subsequently, the T10 vertebra underwent dorsal laminectomy. The 10-g node was allowed to fall freely from the height of 2.5 cm, causing contusion injury to the spinal cord. The muscles and skin were sutured. The hindlimbs of the rats exhibited involuntary spasms and the tails wriggled, indicating that the injury was consistent with the criteria of SCI in this model. Cefuroxime sodium was used for 3 days post-surgery to prevent incision infection. Bladder evacuation was applied twice a day for 7 days post injury. In the sham group, rats received only laminectomy and showed normal Basso, Beattie, and Bresnahan locomotor rating scores post-surgery.
DFO treatment
DFO (100 mg/kg per day; Novartis, Basel, Switzerland; 500 mg dissolved in 5 mL of 0.9% normal saline) was administrated by intraperitoneal injection 30 minutes before injury,and then was injected once a day for 7 days post injury. As a control, the rats in sham and SCI groups were injected with 0.9% NaCl (1 mL/kg). The dosage of DFO administration was determined based on our previous methods (Hao et al.,2017).
Basso, Beattie, and Bresnahan locomotor rating scale
The functional restoration was assessed according to the Basso, Beattie, and Bresnahan locomotor rating scale and assessed by two individual researchers (Basso et al., 1995).The Basso, Beattie, and Bresnahan locomotor rating scores for evaluating hindlimb locomotor function ranged from 0(no observable movement) to 21 (normal movement). Before evaluation, the rats were allowed to move freely for 5 minutes in the open filed. The Basso, Beattie, and Bresnahan locomotor rating scores were determined 0, 1, 2, 4, and 8 weeks post-SCI to assess locomotor recovery.
Determination of iron concentration
Spinal cord samples were collected and homogenized under ice-cold conditions. Iron levels of spinal cords at 1, 4, and 24 hours post-SCI were detected using the tissue iron assay kit (Nanjing Jiancheng Bioengineering Institute, Nanjing,China). To measure iron concentration in the sample, the optical density (OD) value was measured at 520 nm using a spectrophotometer (752-P; Shanghai Xian Ke, Shanghai,China) and the iron concentration in the samples was then determined by comparing the OD of the samples to the standard curve.
Transmission electron microscope
Samples in the sham and SCI groups at 1 and 24 hours of recovery were perfused in 2% paraformaldehyde, 2% glutaraldehyde in 0.1 M sodium cacodylate, fixed in 2% osmium tetroxide with 1.6% potassium ferrocyanide in 0.1 M sodium cacodylate. Samples were then cut into small clumps with a volume of 1 mm3 and stained with 2% uranyl acetate, dehydrated in ethanol, and embedded in Eponate. The sections were placed on copper slot grids and stained with 2% uranyl acetate and lead citrate. Images were captured with a transmission electron microscope (7600; Hitachi, Tokyo, Japan).
Western blot assay
Rats were sacrificed at 2 or 7 days post-SCI to detect the expression of glutathione peroxidase 4 (GPX4, a ferroptosis marker), system Xc-light chain (xCT) (ferroptosis marker),and 4-hydroxynonenal (HNE, a lipid perioxidation marker).Spinal cord epicenters 0.5 cm in size were collected and lysed by homogenization in 300 μL lysis buffer (20 mM Tris pH 7.4, 50mM NaCl, 1% Triton X-100 and protease inhibitor).The homogenates were centrifuged, and the supernatants were collected, aliquoted, and stored at -80°C for later use.The samples were dissolved in loading buffer, boiled for 5 minutes, and centrifuged at 6000 r/min at 4°C for 3 minutes to collect the protein supernatant. The proteins were loaded on gels for electrophoresis and were transferred to polyvinylidene difluoride membranes by a transfer apparatus at 65 V for 2 hours at 4°C. The membranes were blocked at room temperature for 2 hours on the shaker. Sequentially, the membranes were incubated with primary antibodies against GPX4, xCT and HNE at 4°C overnight. After three washes with Tris buffered saline with Tween, each for 10 minutes,goat-anti-rabbit IgG conjugated with horseradish peroxidase (1:2000, SA00001-2; Proteintech Group, Chicago, IL,USA) was added for an additional 1.5 hours. After Tris buff-ered saline-Tween washes, the blots were visualized using an enhanced chemiluminescence system (ChemiDox XRS;Bio-Rad, Hercules, CA, USA). The target protein bands'grayscale values were quantified using ImageJ software (NIH,Bethesda, MD, USA). All grayscale values were normalized to that of β-actin or GAPDH for statistical evaluation. The details of primary antibodies are shown in Table 1.
Glutathione detection
The spinal cord samples were collected as previously described at 2 or 7 days post-SCI, and spinal cord homogenate was prepared under ice cold conditions. Glutathione (GSH)level was detected according to total GSH assay kit (Shanghai Ke Shun, Shanghai, China) instructions. To measure GSH concentration in the sample, the OD value was measured at 450 nm using a multiscan spectrum (Infinite M200 Pro, TECAN, Switzerland) and the GSH concentration in the samples was then determined by comparing the OD of the samples to the standard curve. All assays were performed in triplicate.
Total RNA preparation and real-time quantitative polymerase chain reaction
Spinal cord samples were immediately removed after rats were sacrificed at 1 hour, 8 hours, and 3 days after SCI and frozen on dry ice powder. Spinal cord segments were homogenized in ice-cold Trizol. Chloroform was added, and the aqueous phase was collected after centrifugation. RNA extraction was done according to the manufacturer's protocol (Qiagen RNeasy Mini, Qiagen, Valencia, CA, USA).RNA quality was detected using Agilent Bioanalyzer (Agilent, Palo Alto, CA, USA) in that all samples should show sharp ribosomal RNA bands. One mg of total RNA wasused for first-strand cDNA production with Super Script II reverse transcriptase (Invitrogen, Carlsbad, CA, USA)primed by Oligo dT. The real-time quantitative polymerase chain reaction (RT-qPCR) was performed on an Applied Biosystems 7900HT machine, and 10 ng of cDNA, 50 nM of primers, and SYBR Green master mix (Applied Biosystems,Foster City, CA, USA) was used. Expression levels of each target gene were normalized to the mean critical threshold(CT) values of the β-actin housekeeping gene using the 2-ΔΔCtmethod (Livak and Schmittgen, 2001) to calculate the relative amount of RNA. The primers used are shown in Table 2.
Table 1 Primary antibody details

Table 2 Primers of real-time quantitative polymerase chain reaction
Histology
Rats were anesthetized and perfused with 200 mL cold saline, followed by 300 mL cold 4% paraformaldehyde at 2 and 8 weeks post-SCI. Spinal cord epicenters 1 cm in size were excised and stored in 4% paraformaldehyde at 4°C for 24 hours. Samples were embedded in paraffin and sliced into 5-μm-thick sections. The sections were subjected to hematoxylin-eosin staining according to the procedure described above (Wang et al., 2014a).
Immunofluorescence staining
After being sacrificed at 2 and 8 weeks after SCI, the rat spinal cords were collected, paraffiin spinal cord sections (5 μm thick) from each specimen were deparaffiinized with xylene(2 × 20 minutes), incubated in graded concentrations of ethanol (2 × anhydrous ethanol, 95%, 90%, 80%, and 70%),and then washed in phosphate buffered saline (PBS) for 3 ×3 minutes. Sections were incubated in H2O2for 15 minutes to block endogenous peroxidase. Subsequently, the sections were incubated with a blocking solution (0.1% Triton X-100,10% normal goat serum in PBS) at room temperature for 1 hour. Next, the samples were incubated with the primary antibodies overnight at 4°C. Details of the primary antibodies used are shown in Table 1. Sections were washed for 3 ×3 minutes with PBS, and then incubated with the second antibodies (donkey anti-goat IgG H&LAlexa Fluor® 488, 1:200,ab150129; Abcam, Cambridge, MA, USA; donkey anti-rabbit IgG H&LAlexa Fluor®647, 1:200, ab150075; Abcam) for 60 minutes at room temperature. After three rinses in PBS,the nuclei were counterstained with 1 g/mL DAPI (Biosharp,Hefei, Anhui Province, China) for 5 minutes. Finally, a drop of anti-fade mounting medium (Solarbio, Beijing, China)was placed on each slide, and a coverslip was placed on top of each sample. The immunofluorescence imaging was visualized using a fluorescence microscope (IX71; Olympus,Tokyo, Japan). Neuronal nuclear antigen (NeuN) expression at dorsal horn and glial fibrillary acidic protein (GFAP) expression at the dorsal column lesion were visualized. ImageJ software (NIH) was used to assess the percentages of NeuN and GFAP expression of representative fluorescence micrographs at the injury epicenter.
Statistical analysis
All data are presented as the mean ± SEM. Statistical analysis was performed using GraphPad Prism 5 software (GraphPad Software, Inc., San Diego, CA, USA). Comparisons between multiple groups were made using two-way repeated measures analysis of variance followed by Tukey's post hoc test.A value of P < 0.05 was considered statistically significant.
Results
Effect of DFO on locomotive activity in rat models of SCI
As shown in Figure 1B, severe hindlimb paralysis was observed in the SCI group. Hindlimb locomotive function was improved in DFO-treated SCI rats as early as 7 days post injury (P < 0.05; n = 3; two-way analysis of variance followed by Tukey's post hoc test). Throughout the assessments period of 8 weeks, the DFO group outperformed the SCI group. These data indicated that DFO treatment could promote long-term functional recovery after SCI. Next, we explored the mechanism of DFO as a ferroptosis inhibitor in SCI repair.
Hemolysis, mitochondrial change, and iron overload after SCI
A transmission electron microscope was used to examine the ultrastructure of spinal cords after SCI (Figure 2A).Hemolysis was observed 1 hour post-injury and became more common at 24 hours. However, in the sham group, no hemolysis was observed. Hemolysis results from erythrocyte disruption, which is a source of iron overload after SCI. In both sham and SCI groups, there was no obvious apoptosis morphology at 1 and 24 hours, which is consistent with previous studies that apoptosis occurs at 1-2 days post injury(Nottingham and Springer, 2003). However, as early as 1 hour post injury, we found shrunken mitochondria, occasionally with broken outer membranes in SCI tissues. The sham group showed normal morphology of mitochondria.The ferroptotic-specific change in mitochondria morphology indicates that ferroptosis may occur after SCI.
Next, the iron levels of different study groups were detected at 1, 4, and 24 hours (Figure 2B). Iron level showed an obvious increase in the SCI group compared with the sham group (P < 0.001; n = 3; two-way analysis of variance followed by Tukey's post hoc test), whereas DFO treatment significantly decreased the iron concentration in the DFO group compared with the SCI group (P < 0.001). Therefore,DFO effectively decreased iron levels after SCI. Iron overload is a key factor that triggers ferroptosis. Therefore, DFO may inhibit ferroptosis in the secondary phase of SCI.
Figure 1 Effect of DFO on hindlimb function of rats with SCI.
Ferroptosis pathway in SCI and its inhibition by DFO
At 2 and 7 days post SCI, the xCT expression, which represents Xc- level, was decreased at 7 days post SCI (P < 0.001).This indicated that DFO effectively upregulated xCT expression post-SCI (P < 0.001; Figure 3A and B).
GPX4 plays a central role in ferroptosis (Angeli et al.,2017). Upon SCI, the GPX4 expression significantly decreased from 2 to 7 days post injury (P < 0.05), which indicates that ferroptosis is involved in secondary injury of SCI.DFO effectively upregulated GPX4 expression compared with the SCI group (P < 0.001; Figure 3C and D).
HNE is the product of lipid peroxidation reaction (Girotti,1998). Western blot assay showed that HNE was increased after SCI (P < 0.01) and was markedly decreased in the DFO group (P < 0.001; Figure 3E and F). This result was consistent with the altered expression of GPX4, which directly removes lipid reactive oxygen species.
Tripeptide GSH is a critical linker in the ferroptosis pathway. Cysteine is provided by xCT as a precursor for the synthesis of GSH. GSH maintains the normal function of GPX4 by reducing GPX4 and releasing it from the membrane to the cytosol (Cozza et al., 2017). GSH levels reduced after SCI, and DFO treatment robustly upregulated GSH (P <0.001; Figure 3G).

Figure 2 Iron overload after SCI and transmission electron microscopy of hemolysis and ferroptosis-specific mitochondrial changes.
Collectively, major factors in ferroptosis pathway, including the system Xc-/GSH/GPX4/HNE axis, are associated with secondary injury of SCI. We found that DFO inhibits the ferroptosis pathway by upregulating the Xc-/GPX4 axis and inhibiting GSH depletion and lipid reactive oxygen species (Figure 3H).
DFO effects on ferroptotic-specific mitochondria genes in SCI
The mRNA levels of two ferroptotic related genes, ACSF2 and IREB2 genes, at 1 hour, 8 hours, and 3 days post-injury are shown in Figure 4. The mRNA levels of IREB2 and ACSF2 in the SCI group were increased compared with the sham group at 1 and 8 hours post injury (P < 0.05). DFO treatment significantly decreased IREB2 and ACSF2 mRNA expression compared with the SCI group (P < 0.05).
DFO effects on spinal cord tissue sparing
Figure 3 Expression of ferroptosis-regulator xCT,GPX4, GSH, and HNE.
Spinal cord sections revealed marked tissue sparing follow-ing DFO treatment compared with the SCI group, as shown in representative hematoxylin-eosin staining micrographs.The spinal cord tissue sample from the sham group was intact. DFO treatment therefore remarkably reduced secondary injury post SCI, as shown by increasing spared tissue,which was associated with improved hindlimb functional recovery (Figure 5).
DFO treatment increases the number of NeuN+ cells after SCI
To identify which neuronal cell type reacts to DFO treatment, we examined the effect of DFO on neurons. The number of NeuN+cells in the SCI group was decreased at 2 and 8 weeks after injury compared with the sham group (P < 0.001;Figure 6). However, DFO increased the number of NeuN+(neuronal nuclear antigen) cells (P < 0.001), which suggests that DFO treatment can protect neurons. Therefore, DFO has obvious effects on neuronal preservation.
DFO treatment decreases GFAP expression after SCI
To determine whether DFO treatment affects glial cells after SCI, we examined the effect of DFO on activated astrocytes.GFAP is a marker of activated astrocytes. The number of GFAP+astrocytes in the SCI group was larger than that in the sham group, which is indicative of robust gliosis at 8 weeks post injury (P < 0.001; Figure 7). However, there were slightly more GFAP+astrocytes in the DFO group than in the Sham group. This effect was more obvious at 8 weeks post injury. DFO significantly reduced gliosis (P < 0.001),which indicates that DFO did not increase scar tissue but preserved normal tissue. The effect of anti-gliosis of DFO was consistent with its effect on up-regulating GPX4. GPX4 ablation also induced severe gliosis.
Discussion
Neuronal cell death is a major event of SCI secondary injury (Huang et al., 2015; Liu et al., 2016). Although apoptosis is widely investigated in the pathogenesis of acute SCI, the caspase inhibitor, which limits apoptosis, has shown no benefits in SCI repair (Ozawa et al., 2002). The therapeutic strategies targeting currently known cell death pathways are largely ineffective. Our data indicate that ferroptosis provides an alternative interpretation of SCI secondary cell death. Our results demonstrated that, after SCI, mitochondria in the damaged tissue showed ferroptotic morphology, and molecules of the ferroptosis pathway (xCT, GPX4, GSH, 4HNE)were altered after injury and rescued by DFO treatment. We also detected the expression profile of ferroptosis-specific mitochondria genes.
Ferroptosis is correlated with secondary injury of SCI
In recent years, accumulating studies have reported the role of ferroptosis in the pathogenesis of neurodegenerative diseases. So far, the role of ferroptosis in SCI is unknown.Because the metabolism of iron and lipid is associated with SCI pathophysiology, we speculated that ferroptosis plays a key role in secondary injury of traumatic SCI.
Figure 4 Effects of DFO on ferroptosis specific genes expression at 1 hour, 8 hours, and 3 days after SCI.
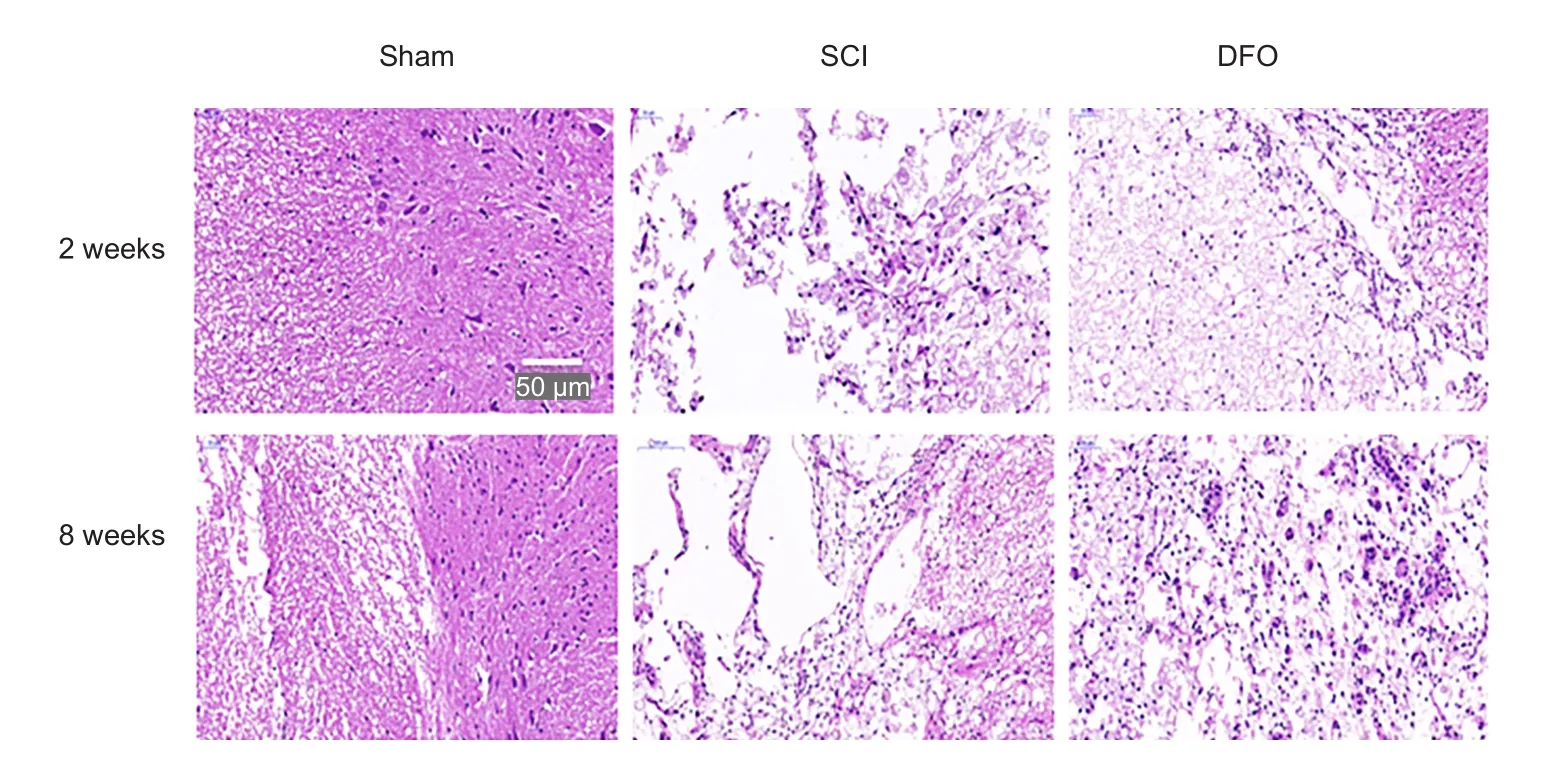
Figure 5 Effect of DFO treatment on histology following SCI.
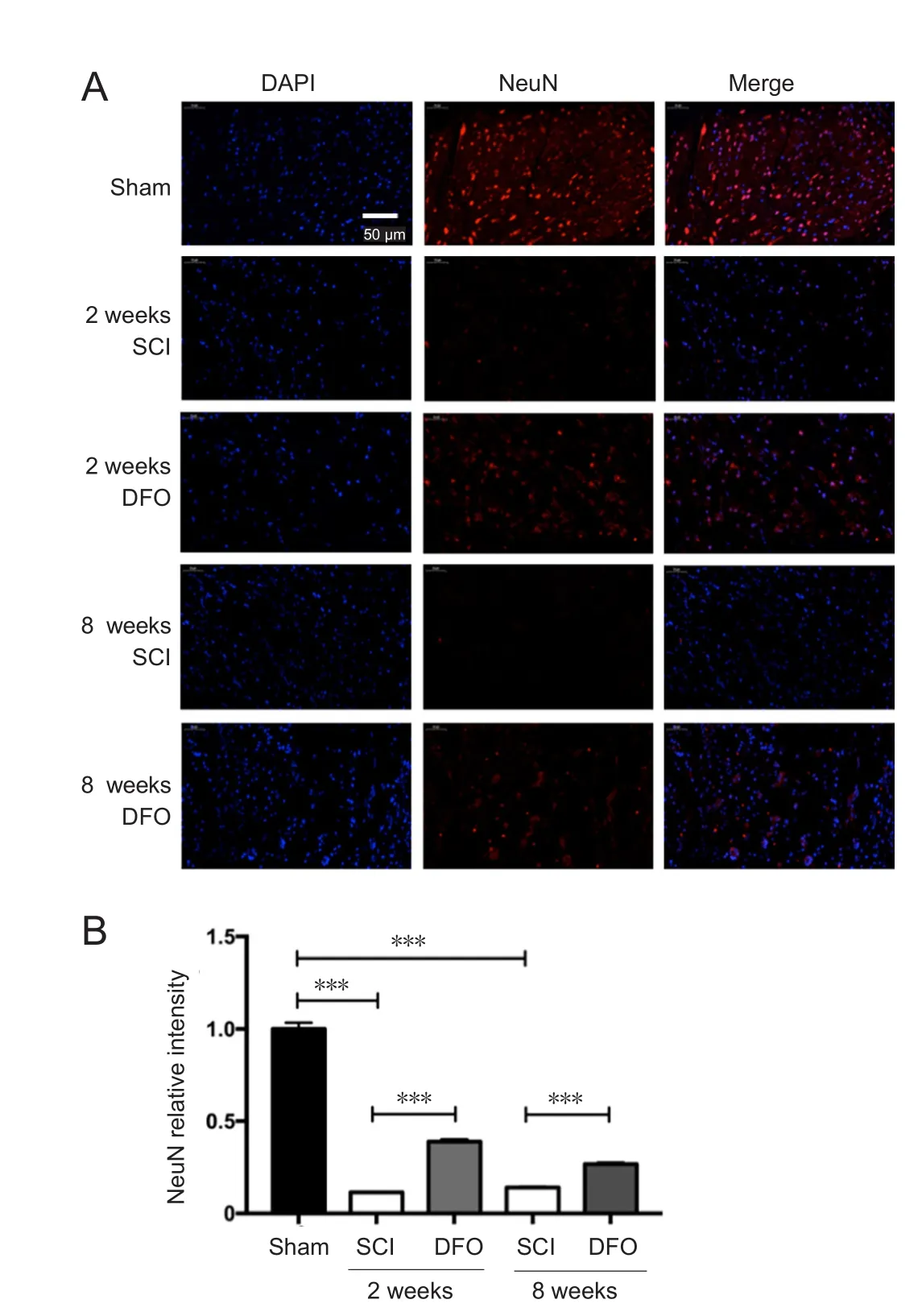
Figure 6 Effects of DFO on NeuN immunofluorescence at 2 and 8 weeks after SCI.
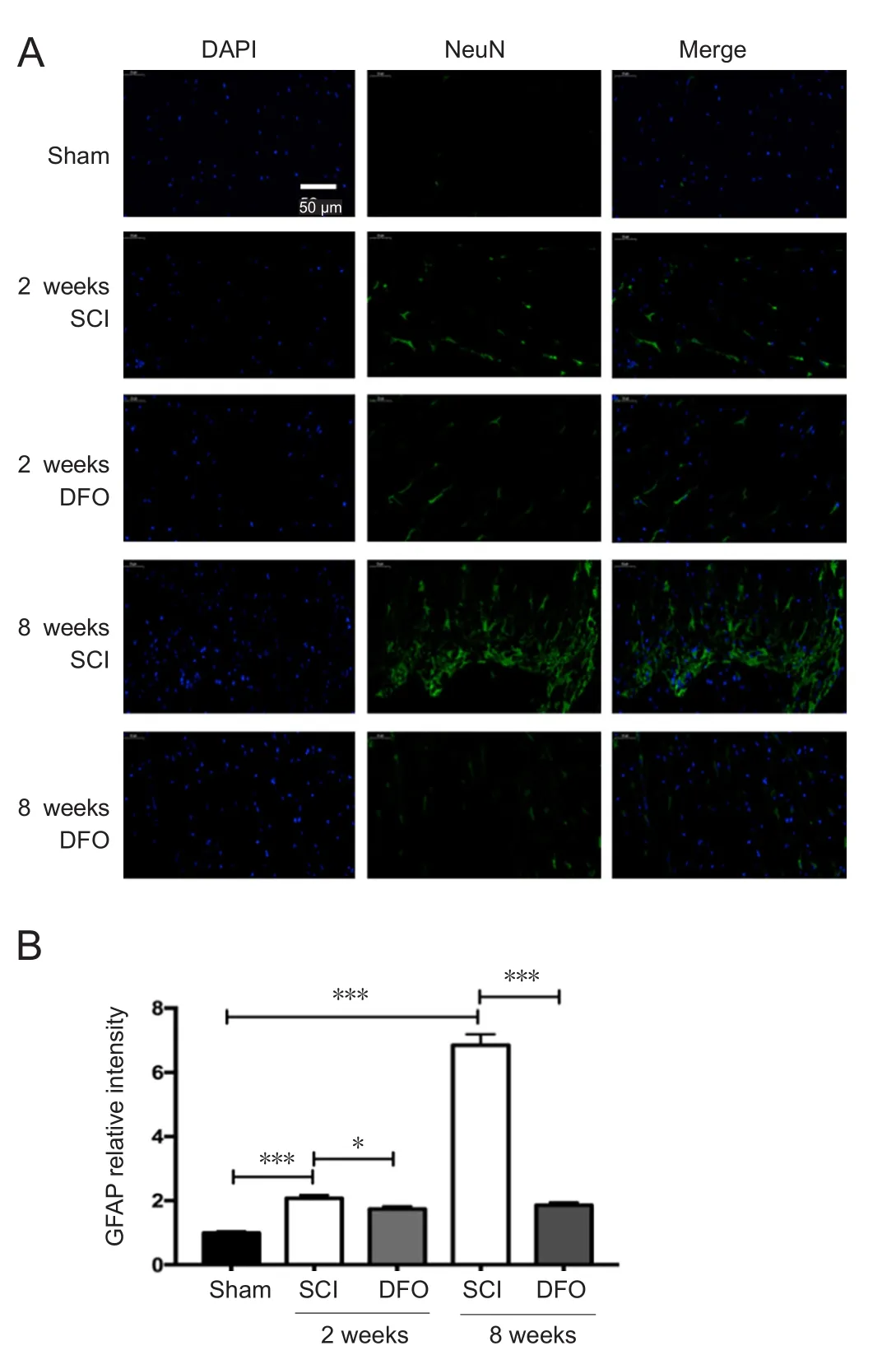
Figure 7 Effects of DFO on GFAP immunofluorescence at 2 and 8 weeks after injury.
In this study, the key factors in the ferroptosis pathway were involved in SCI pathology. The expression of GPX4 and xCT was down-regulated upon SCI. We found that DFO treatment upregulated the Xc-/GPX4 axis, reducing GSH depletion and lipid perioxidation. However, further research is required to determine the spatial and temporal patterns of key ferroptosis regulators such as GPX4 in the secondary phase of SCI.
We observed ferroptotic mitochondrial changes upon SCI as decreased ridges, ruptured outer membranes, and reduced size. These data strongly support the view that ferroptosis occurs in damaged spinal cord tissue. We further explored the expression pattern of mitochondrial genes RPL8, IREB2,CS, ATP5G3, TTC35, and ACSF2, which have been identified as ferroptosis-specific genes. Inhibition of these six genes by siRNA reduces ferroptosis (Dixon et al., 2012). We examined all six genes and identified two (ACSF2 and IREB2) that were SCI-relevant. No significant changes in CS, PRL8, TTC35,or ATP5G3 mRNA levels were found at 1 hour, 8 hours, or 3 days post SCI, whereas IREB2 and ACSF2 showed remarkable upregulation at 1 hour and 8 hours post SCI. The pathological processes of different diseases may have different expression profiles. Involvement of IREB2 further implicates the role of iron regulation in the pathogenesis of SCI. ACSF2 is associated with lipid metabolism (Watkins et al., 2007). We found IREB2 and ACSF2 changes in the acute phase of SCI, and that DFO downregulated these two genes. Further study is needed to understand the role of these two genes in the pathophysiological process of SCI.
Mechanism of DFO in SCI repair is through ferroptosis inhibition
The effectiveness of DFO in SCI has received increasing attention. DFO is a safe drug and has been reported to be effective in rat models of SCI. Its mechanism has been reported as reducing iron overload, reactive oxygen species production, inflammation, and gliosis. However, it remains unclear why iron chelation induces obvious regeneration.Translating DFO into a therapeutic application will require an understanding of how DFO works to promote tissue regeneration. The efficacy and safety of the iron chelator,deferiprone, have also been investigated in a clinical trial for Parkinson's disease treatment (Martin-Bastida et al.,2017a, b).
Iron chelation by DFO can rescue ferroptosis induced by erastin in cancer cells (Dixon et al., 2012). A previous study found that the spinal cord had obviously intensified staining for GFAP+astrocytes in Gpx4NIKO mice, as well as less staining for NeuN+cells (Chen et al., 2015). Here, we reported an increase in GPX4 expression in DFO treatment. The role of DFO on neuronal protection and gliosis reduction could be interpreted as the effect of upregulation of GPX4,the key regulator of ferroptosis.
In the DFO group, the expression levels of xCT, GSH, and GPX4 were much higher than in the sham group; however,DFO treatment did not increase the levels of these factors in uninjured tissues (unpublished data). The mechanism of such robust upregulation requires further study.
DFO administration was given 30 minutes before SCI, because we aimed to increase plasma levels of DFO to prohibit iron overload post SCI. This administration is consistent with a previous report (Paterniti et al., 2010). The effectiveness of DFO treatment identified ferroptosis as a novel therapeutic target for SCI treatment, although the time window needs further study.
In summary, DFO improved recovery of SCI by inhibition of the ferroptosis pathway in a contusion model of SCI. Our study revealed the role of ferroptosis in secondary injury of SCI and should encourage the development of novel therapeutics that specifically target ferroptosis.
Acknowledgments:Prof. Wise Young from Keck Center for Collaborative Neuroscience Nelson Biological Laboratories, Rutgers, the State University of New Jersey also provide valuable suggestions on the results writing. We also thank Prof. Fabin Han from Taishan Medical University for giving critical review of this paper.
Author contributions: Study design and concept: SQF, XY, XHK and JH; experiment implementation: YZ, HQD, CS and CXZ; provision of critical reagents and scientific input: CL and BL; rat management: CXZ,XW, WXL, XHF and BYF; data analysis and paper preparation: YZ, JH and XY. All authors approved the final version of the paper.
Conflicts of interest:The authors declare that there are no conflicts of interest associated with this manuscript.
Financial support: This study was supported by the National Natural Science Foundation of China, No. 81672171 (to XY), 81330042 (to SQF),81620108018 (to SQF), 81772342; the State Key Laboratory of Medicinal Chemical Biology (Nankai University), China, No. 2017027. The funding sources had no role in study conception and design, data analysis or interpretation, paper writing or deciding to submit this paper for publication.
Institutional review board statement: All experimental procedures and protocols were approved by Animal Care and Research Committee of Institute of Radiation Medicine Chinese Academy of Medical Sciences,Tianjin, China (approval No. DWLI-20171010). All experimental procedures described here were in accordance with the National Institutes of Health (NIH) guidelines for the Care and Use of Laboratory Animals.
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement:Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix, tweak,and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewers: Tania Cristina Leite de Sampaio e Spohr, Instituto Estadual do Cérebro Paulo Niemeyer, Laboratorio de Biomedicina do Cérebro, Brazil; He Huang, Central South University Xiangya School of Medicine, China.
Additional file:Open peer review reports 1 and 2.
杂志排行

中国神经再生研究(英文版)的其它文章
- The role of Epstein-Barr virus in multiple sclerosis:from molecular pathophysiology to in vivo imaging
- The metabolome identity: basis for discovery of biomarkers in neurodegeneration
- Neuroinflammation as a target for glaucoma therapy
- Basics on the use of acid-sensing ion channels'inhibitors as therapeutics
- Role of axon resealing in retrograde neuronal death and regeneration after spinal cord injury
- Rehabilitation following spinal cord injury: how animal models can help our understanding of exerciseinduced neuroplasticity