Role of axon resealing in retrograde neuronal death and regeneration after spinal cord injury
2019-02-13WilliamRodemerMichaelSelzer
William Rodemer, Michael E. Selzer,
1 Shriners Hospital Pediatric Research Center (Center for Neural Repair and Rehabilitation), Philadelphia, PA, USA
2 Department of Neurology, the Lewis Katz School of Medicine at Temple University, Philadelphia, PA, USA
Abstract Spinal cord injury leads to persistent behavioral deficits because mammalian central nervous system axons fail to regenerate. A neuron's response to axon injury results from a complex interplay of neuron-intrinsic and environmental factors. The contribution of axotomy to the death of neurons in spinal cord injury is controversial because very remote axotomy is unlikely to result in neuronal death, whereas death of neurons near an injury may reflect environmental factors such as ischemia and inflammation. In lampreys,axotomy due to spinal cord injury results in delayed apoptosis of spinal-projecting neurons in the brain,beyond the extent of these environmental factors. This retrograde apoptosis correlates with delayed resealing of the axon, and can be reversed by inducing rapid membrane resealing with polyethylene glycol.Studies in mammals also suggest that polyethylene glycol may be neuroprotective, although the mechanism(s) remain unclear. This review examines the early, mechanical, responses to axon injury in both mammals and lampreys, and the potential of polyethylene glycol to reduce injury-induced pathology. Identifying the mechanisms underlying a neuron's response to axotomy will potentially reveal new therapeutic targets to enhance regeneration and functional recovery in humans with spinal cord injury.
Key Words: axon resealing; regeneration; retrograde neuronal death; spinal cord injury; sea lamprey; PEG;mitochondrial dysfunction; calcium signaling
Introduction
Immediately after neural trauma, severed axons must reseal their axolemma to restore homeostasis and limit exposure to toxic-mediators in the extracellular environment. In mammals, this process often ends in the formation of dystrophic end bulbs with accompanying loss of function (Ramón y Cajal and May, 1928). Unlike mammals, the sea lamprey, Petromyzon marinus, recovers swimming behavior after complete spinal cord transection facilitated by heterogeneous regeneration of the reticulospinal (RS) system. Those neurons that fail to regenerate undergo a delayed form of apoptosis (Shifman et al., 2008). In a recent article in Brain Sciences, we demonstrated that artificially inducing membrane resealing with polyethylene glycol (PEG) reduces injury-induced caspase activation within this population (Zhang et al., 2018a). The present review leans heavily on the lamprey findings, but PEG has also shown potential therapeutic benefits in mammalian axon injury. Although the underlying mechanism(s)are still not fully understood, it is clear that the determinants of axon regeneration and cell survival are strongly influenced by the initial cellular response to transection-induced trauma.Elucidating the key steps in activating the regeneration program or initializing a terminal caspase cascade will yield new insights and reveal novel molecular targets to promote neural regeneration after traumatic injury, not only in lampreys, but also in mammals. We have performed a PubMed literature search of articles on axotomy-induced neuronal death, axon resealing, and PEG as a therapeutic fusogen published from January 1989 to September 2018.
Neuron Survival after Axon Injury
In mammals, the question of whether spinal cord injury induces retrograde neuronal of death of spinal projecting neurons remains controversial. Following transection of T9 dorsal corticospinal tract (CST) fibers, 39% of retrogradely labeled rat corticospinal neurons became terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)positive within 1 week of injury, which was followed by an approximate 50% reduction in cell count between 2 and 4 weeks, suggesting significant cell death. Similar findings have been observed in rats among retrogradely labeled rubrospinal neurons after cervical spinal cord injury, and within the pontine reticular formation after complete T8 transection(Mori et al., 1997; Novikova et al., 2000; Wu et al., 2003).However, these studies have been challenged by reports that suggest that spinal cord injury induces atrophy, rather than cell death, among spinal projecting neurons. These studies did not detect significant supraspinal cell loss but found that surviving neurons were significantly smaller in size than uninjured controls (McBride et al., 1989; Nielson et al., 2011).In one notable study, a stereological analysis in rat red nucleus and motor cortex found no significant reduction in surviving neurons 1 year after unilateral transection of the C4 dorsolateral funiculus (Kwon et al., 2002). However, the lack of retrograde labeling in this study prevented a clear determination that all of the cells included in the analysis were truly injured.
The most plausible explanation for the discrepancy of atrophy vs. death is that retrograde neuronal death is partly a function of the distance of the axotomy from the neuron's cell body. When axotomy is close to the cell body, as in optic nerve crush, neuron death is unambiguous. After optic nerve crush, over 90% of rat and approximately 73% of mouse retinal ganglion cells undergo apoptosis (Garcia-Valenzuela et al., 1994; Mead et al., 2014). In these models, suppressing the caspase cascade after optic nerve crush by overexpressing the anti-apoptotic bcl2 family protein, Bcl2, or inhibiting the pro-apoptotic bcl2 protein, Bax, provided neuroprotection(Chierzi et al., 1998; Qin et al., 2004). Similarly, around half of CST neurons are lost by two weeks after subcortical axotomy (Hollis et al., 2009). Within the spinal cord, local cell death occurs around the injury site and retrograde tracing has shown that propriospinal neurons are selectively vulnerable to axotomy close to the cell body (Conta Steencken et al., 2011). However, the closer an injury is to the cell body,the more likely it is that environmental factors such as ischemia and inflammation can account for the neuronal death.While it is clear that axotomy-induced retrograde cell death occurs in mammals under certain conditions, the diffiiculty in obtaining unambiguous results and untangling the multifaceted mechanisms driving these processes makes a strong argument for modeling spinal cord injury in simpler nervous systems.
One of the major advantages of the lamprey model is the lack of ambiguity in the spinalized nervous system. Supraspinal motor control is mediated by the RS system, which comprises approximately 2000 small neurons along with 18 pairs of large identifiable neurons (McClellan, 1992; Swain et al., 1993; McClellan, 1994). After complete transection,approximately 50% of the lamprey's large identifiable RS neurons regenerate their axons across the lesion (Davis and McClellan, 1994). Many of those large neurons that fail to regenerate ultimately die by a delayed form of caspase-mediated apoptosis. Studies have detected active caspases in the proximal axon tip by 1 day after transection and in the brain as early as 1 week post-injury (Barreiro-Iglesias and Shifman,2012; Hu et al., 2013; Barreiro-Iglesias and Shifman, 2015).Despite early caspase activation, TUNEL-positive staining has not been reported until approximately 1 month after injury and cells do not begin to disappear until 4 months(Shifman et al., 2008). Since caspase activation occurs long before any possible anatomical regeneration, the delayed apoptosis observed in poorly regenerating RS neurons is likely initiated as a primary physiological response to transection and not as a secondary consequence of failing to regenerate. Although the long delay between caspase activation and cell death presents a window to rescue poorly regenerating neurons, the best opportunity for therapeutic intervention may be immediately after injury.
Axon Resealing Kinetics
Traumatic axon injury depolarizes the membrane and leads to an influx of cations and other components of the extracellular milieu into the axoplasm. Calcium entry, in particular,is essential to initiate the membrane repair process, which involves vesiculation of adjacent axolemma to form a plug.Molecularly, this process depends on intracellular cascades that involve local activation of calpains, increased levels of cAMP, and cytosolic oxidation, which ultimately lead to activation of the vesicle recycling protein, N-ethylmaleimide-sensitive factor (Howard et al., 1999; Zuzek et al., 2013).The kinetics of axon repair vary widely among species, neuron types, and distance of the injury from the soma. Among invertebrates, reported resealing times after transection include: less than 10 minutes for cultured sea slug (Aplysia californica) axons, less than 30 minutes for the cockroach(Periplaneta american) ventral nerve, 1 hour for earthworm(Lumbricus terrestris) giant axons, and more than 2.5 hours for axons from the giant squids (Loligo paelei and Sepioteuthis lessoniana) (Yawo and Kuno, 1985; Spira et al., 1993;Krause et al., 1994). It is important to note that even after a functional seal is achieved, the repair process continues as vesicles form an increasingly tighter and more stable plug. In earthworms, morphological assays showed that the complete process can take over 24 hours post-transection(Lichstein et al., 2000). Axon resealing has not been examined as intensively in the mammalian nervous system, due to its complexity. In isolated white matter strips from guinea pig (Cavia porcellus) spinal cord, axons sealed to horseradish peroxidase (HRP; 44 kDa, 308 amino acid residues) within 30 minutes of transection (Shi et al., 2000). Over 80% of mouse B104 neuroblastoma cells axotomized more than 50 μm from the perikaryon resealed within 10 minutes, compared to only 50% of cells transected less than 50 μm from the cell body (McGill et al., 2016). For both the guinea pig explant and the B104 cells, axon resealing was dependent on the presence of calcium in the culture medium as resealing was inhibited in low calcium concentrations.
In lampreys, the resealing time of an RS axon is closely correlated with the cross-sectional area of its perikaryon, which in turn correlates with the caliber of its axon (Zhang et al.,2018a). Axons of the largest neurons remained open to fluorescently-tagged 10,000 MW dextrans in excess of 24 hours,albeit with reduced labeling intensity, indicating a partial seal. Intriguingly, this is consistent with the observation of a strong, inverse correlation between lamprey neuron size and the probability that its axon would regenerate (r = -0.92, P< 0.0001, unpublished observations), which suggests that delayed resealing might be an important factor inhibiting axon regeneration. Critically, over 60% of RS neurons with axons that remained open for at least 24 hours were positive for activated caspases at 2 weeks after transection, compared with less than 10% of neurons with sealed axons. Together, these results indicate that axon resealing after transection may play a critical role in determining cell fate (Figure 1).
Axotomy-Induced Mitochondrial Dysfunction
Traumatic axotomy exposes the interior of the cell to the extracellular environment leading to a precipitous influx of cations and, potentially, other toxic factors. After injury, in both lampreys and mammals, free cytosolic calcium rises well above physiological ranges forming a spatiotemporal gradient that is maximal at the injured tip (Strautman et al.,1990; Ziv and Spira, 1995). Moreover, injury-induced membrane depolarization, calpain activation, and high levels of free cytosolic calcium and sodium lead to a second influx of: a) extracellular calcium through voltage-gated calcium channels and reversal of the sodium-calcium exchanger; and b) release of calcium from intracellular stores (Stys, 2005;Villegas et al., 2014). Both sources of calcium are buffered,in part, by calcium binding proteins in the cytosol, such as parvalbumin, and by local mitochondria, which remove calcium from the cytosol principally through the mitochondrial calcium uniporter (Ganitkevich, 2003; Obal et al., 2006).However, high levels of calcium can overwhelm the buffering capacity of mitochondria, increasing oxidative stress and leading to the opening of the permeability transition pore on the mitochondrial inner membrane (Barrientos et al., 2011).This, in turn, leads to mitochondrial swelling, the generation of reactive oxygen species, adenosine triphosphate depletion,cytochrome c release, and release of mitochondrial calcium into the cytosol. Inhibiting either the influx of extracellular calcium or release of calcium from intracellular stores can be neuroprotective (Stys et al., 1990; Stys, 2005). However, chelating extracellular calcium alone is not suffiicient to prevent mitochondrial dysfunction after membrane injury (Villegas et al., 2014).
In lampreys, removing calcium from the dissecting fluid and chelating extracellular calcium with ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid(EGTA) not only prolonged the time to axolemmal resealing,but also exacerbated caspase activation (Zhang et al., 2018a).These results indicated that the initial influx of extracellular calcium was not the primary determinant of injury-induced degeneration in lamprey neurons. Rather, caspase activation had to be a consequence either of influx of other toxic factors from the extracellular environment, or of secondary calcium entry into the cytosol from intracellular stores, or across the axolemma, once the EGTA was washed out. Massive entry of sodium while the axolemma is still unsealed might lead to reversal of the Sodium-calcium exchanger with net release of calcium from mitochondria and other intracellular organelles, even in the total absence of extracellular calcium. It is possible that mitochondrial dysfunction underlies the cell death observed among large, slowly resealing, RS neurons. Conversely, reduced mitochondrial damage among quickly resealing, small RS neurons may be a key driver of their superior survival and regenerative ability. Thus it is possible that in lamprey axons, in addition to accelerating axon resealing, PEG treatment suppressed apoptosis by exerting a protective effect on mitochondria.
This study is supported by a recent report that gamma-aminobutyric acid (GABA) agonism is neuroprotective in lampreys (Romaus-Sanjurjo et al., 2018). In that study, bath administration of GABA or the GABA-B receptor agonist,baclofen, reduced caspase activation among the large RS neurons at 2 weeks post-transection and, after substituting GABA with the GABA analogue, gamma-amino-β-hydroxybutyric acid (GABOB), promoted axon regeneration at 12 weeks post-injury. Since GABA inhibits calcium influx by inactivating voltage dependent calcium channels, these results suggest that calcium-mediated injury plays an important role in transection-induced retrograde neuronal death. Indeed, the authors noted that in identified RS neurons, increased expression of the GABA-B1 receptor subunit after transection may help compensate for transection-induced calcium influx. Further studies will be needed to determine more precisely the role of calcium after transection and, in particular, the role of mitochondria in guiding the cell towards either regeneration or retrograde neuronal death.
Convergence of Neuron Survival and Axon Regeneration Pathways
Proper mitochondrial function underlies not only cell survival but axon regeneration as well. Mitochondrial translocation to injured C. elegans' axons was an important mediator of regeneration independently of their ability to buffer calcium (Han et al., 2016). In mammals, the extent of neuron death following axon damage depends largely on the severity of injury (Fehlings and Tator, 1995). In lampreys,the strong correlation between the ability of an identified RS neuron to regenerate its axon post-transection and its ability to withstand apoptosis (Shifman et al., 2008) is not a trivial consequence of the inability of dead neurons to regenerate because the apoptosis is delayed for several weeks after regeneration is observed. Rather, neuron-intrinsic factors that determine a neuron's susceptibility to retrograde cell death must represent points of convergence between the signaling for regeneration and for survival after axotomy.
The pathways connecting axon-injury and retrograde neuronal death have yet to be fully elucidated. In mice,particularly in retinal ganglion cells after optic nerve crush,activation of the mTOR pathway, most famously through PTEN deletion, promotes both neuronal survival and axon regeneration (Park et al., 2008). There are at least 30 types of retinal ganglion cells, and most of them die after their axons are crushed. However, the α-retinal ganglion cells selectively survive, and in mice with PTEN deletion, it is these same α-retinal ganglion cells that give rise to the regenerating axons (Duan et al., 2015). These α-retinal ganglion cells have uniquely high levels of mTOR activity, and also selectively express osteopontin and insulin-like growth factor 1.Whether this is true of lamprey RS neurons that are good regenerators and survivors is not yet known, but our recent observations that regenerative ability and likelihood of survival correlate inversely with neuron size and very slow axonal resealing after axotomy suggest that fairly simple mechanical factors may play a role. Moreover, these results suggest that promoting efficient resealing may be a potent strategy to reduce cell death and promote regeneration.
Therapeutic Potential of Polyethylene Glycol
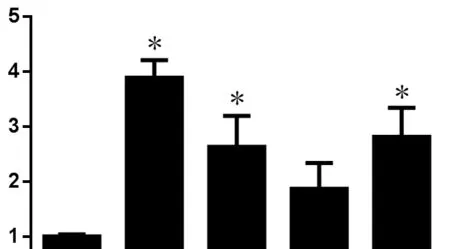
Figure 1 Polyethylene glycol (PEG)-induced axon sealing reduces post-complete spinal cord transection (TX) caspase activation.
The hydrophilic polymer, PEG, is perhaps the most documented membrane fusogen, and is commonly used to generate hybridomas for monoclonal antibody production.Applied to an injured axon, it rapidly induces resealing of damaged plasmalemma, independently of endogenous repair mechanisms, as demonstrated by several studies. For example, in a compression injury model of isolated guinea pig spinal cord explants, application of a 50% w/v solution of 1800 MW PEG restored compound action potentials and significantly reduced horseradish peroxidase (HRP) penetration into injured axons (Shi and Borgens, 1999, 2000). Importantly, PEG sealed the axons effectively regardless of caliber (Shi and Borgens, 2000). In both B104 cultures and rat spinal cord injuryatic nerve explants, PEG induced axolemmal resealing,even in the presence of calcium-free extracellular medium,which inhibits endogenous resealing. Moreover, in B104 cells,PEG was effective at restoring resealing in the presence of antagonistic pharmacological agents that inhibit vesicle mobilization, including the N-ethylmaleimide-sensitive factor inhibitors, N-ethylmalemide and botulinum neurotoxin type A, along with the protein kinase A (PKA) pathway inhibitor,protein kinase inhibitor. Although the exact mechanism has yet to be fully elucidated, the ability of PEG to bypass the endogenous repair process led the authors to speculate that PEG induces fusion of the plasmalemma leaflets directly at the severed stump (Spaeth et al., 2012).
In mammals, the therapeutic potential of PEG has been extensively investigated in a T10-11 compression spinal cord injury model in Guinea pigs. Here, immediate application of a 50% w/v solution of 2000 MW PEG has been shown to reduce oxidative stress and local apoptotic cell death within 1 week of injury (Luo and Shi, 2007). Surprisingly, in this same model, delayed PEG application, even at timepoints as late as 1 month post-injury, has been reported to improve functional outcome and reduce the extent of the lesion (Borgens et al., 2002; Duerstock and Borgens, 2002). In rats, acute IV infusion of a 30% w/v solution of PEG, 2 grams per kilogram total dose, after a T4 compression injury resulted in significantly greater recovery on the BBB scale than controls and,when combined with magnesium sulfate, resulted in greater myelin sparing (Ditor et al., 2007).
These findings suggest that PEG conveys neuroprotection beyond mechanical membrane resealing. It has been reported that PEG protects mitochondria from calcium-mediated damage, possibly by blocking the mitochondria permeability transition pore (Chen et al., 2009). This is supported by additional studies showing mitoprotective effects, including inhibition of oxidative stress and cytochrome c release (Luo et al., 2002, 2004). Consequently, a duel mechanism model of PEG-mediated neuroprotection after axon injury has been proposed. In this model, while most of the PEG applied directly after axon injury acts to reseal the axolemma, some fraction of the polymer enters the axoplasm and interacts with mitochondria to prevent permeability transition. This hypothesis has been supported by in vitro studies using fluorescently decorated PEG to show that PEG can indeed enter the cytosol of damaged neurons (Liu-Snyder et al., 2007).
In addition to membrane resealing, PEG has been a key component of the controversial therapeutic strategy of axon fusion, whereby proximal and distal nerve segments are rejoined to form a continuous functional unit. Although PEG is well known to mediate fusion of cells in culture, most notably in the production of hybridomas used to synthesize monoclonal antibodies, it is less clear whether mammalian axons are intrinsically capable of rejoining once severed in vivo (Davidson and Gerald, 1977). Therapeutic axon fusion is supported, in part, by evidence from invertebrates, including the nematode, Caenorhabditis elegans, where axon fusion occurs as part of the endogenous neural regeneration program(Neumann et al., 2011). In explants of the earthworm ventral nerve cord, PEG application facilitated physical rejoining of apposed nerve segments after transection, and subsequent labeling with Lucifer Yellow indicated morphological continuity of severed giant axons (Krause and Bittner, 1990).Building on earlier work, a 1999 report suggested PEG-mediated axon fusion after Transection could restore electrical conduction in isolated earthworm giant axons, rat spinal cord injuryatic nerve, and rat white matter spinal cord strips. Using a PEG-infused hydrogel, to add mechanical strength, this approach was also able to fuse earthworm giant axons functionally in vivo (Bittner et al., 2015).
Claims regarding PEG's ability to fuse axons in mammals vary widely from modest restoration of electrical conduction after axotomy to facilitating partial motor recovery following complete spinal cord transection (Bamba et al., 2017;Liu et al., 2018). Undoubtedly, strong claims should be considered with caution. In the rat peripheral nervous system,PEG-infused nerve allografts and direct PEG-induced fusion of apposing nerve segments have been reported to facilitate rapid connection across a transection, leading to enhanced plasticity, reduced Wallerian degeneration, reduced loss of muscle innervation, and ultimately, improved functional outcome (Mikesh et al., 2018a, b). These and similar studies have yielded a clinical trial, with results expected in 2019, to assess the ability of 3350 MW PEG to improve nerve function in an interposition graft following traumatic upper extremity nerve injury (Clinical trials identifier,NCT02359825). While this study may give some indication of PEG's potential as a therapeutic fusogen additional studies are needed to fully elucidate the multiple mechanisms by which PEG may convey neuroprotection.
In lampreys, application of a 40% w/v solution of 3350 MW PEG to the proximal spinal cord stump immediately after Transection significantly reduced the number of large RS neurons that remained unsealed 24 hours after injury by approximately 40% (Zhang et al., 2018a). Histological evidence suggested that PEG-induced resealing of the proximal stump rather than fusion between opposing axon segments.As predicted, PEG treatment reduced caspase activation within the perikaryon at 2 weeks post-transection by 69.5%.Because the perikarya of RS axons are more than 1 cm from the transection, they were remote from the local inflammatory response, and thus the neuroprotection was unlikely to be mediated by cell non-autonomous effects (Zhang et al.,2018b). Rather, these results suggest that a delay in resealing after axotomy promotes an intracellular degenerative process that inhibits axon regeneration and ultimately leads to retrograde cell death. Future studies will be needed to elucidate the role of delayed axon resealing on axon regeneration and whether the mitochondrial response to transection differs between those RS neurons that regenerate and those that undergo apoptosis. Identifying the mechanisms connecting axon resealing with regeneration and survival will increase our understanding of traumatic axotomy and potentially reveal new therapeutic targets to enhance recovery in central nervous system-injured humans.
Acknowledgments:The authors thank Kathy Zhang, PhD (Shriners Hospital Pediatric Research Center (Center for Neural Repair and Rehabilitation), Philadelphia, PA, USA) for providing the lamprey micrographs shown in Figure 1 A-D.
Author contributions:Manuscript drafting and writing: WR and MES.
Conflicts of interest: The authors declare no conflicts of interest.
Financial support:This work was supported by grants R01-NS092876 (NIH,to MES), SHC-85400 (Shriners Research Foundation, to MES), SHC-85220(Shriners Research Foundation, to MES).
Copyright license agreement: The Copyright License Agreement has been signed by both authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
杂志排行

中国神经再生研究(英文版)的其它文章
- The role of Epstein-Barr virus in multiple sclerosis:from molecular pathophysiology to in vivo imaging
- The metabolome identity: basis for discovery of biomarkers in neurodegeneration
- Neuroinflammation as a target for glaucoma therapy
- Basics on the use of acid-sensing ion channels'inhibitors as therapeutics
- Rehabilitation following spinal cord injury: how animal models can help our understanding of exerciseinduced neuroplasticity
- The pig as a preclinical traumatic brain injury model:current models, functional outcome measures, and translational detection strategies