亲体肝移植治疗新生儿肝内胆汗淤积型希特林缺乏症1例
2019-01-30刘源夏强张建军薛峰夏雷罗毅邱必军封明轩陈小松韩龙志上海交通大学医学院附属仁济医院肝脏外科上海200127
刘源,夏强,张建军,薛峰,夏雷,罗毅,邱必军,封明轩,陈小松,韩龙志(上海交通大学医学院附属仁济医院肝脏外科,上海 200127)
希特林蛋白缺乏症是由SLC25A13突变导致的一种常染色体隐性遗传病。希特林蛋白(citrin)是一种线粒体内的钙结合天冬氨酸/谷氨酸载体(calcium-stimulated aspartate/ glutamate carrier,AGC)蛋白,在尿素循环及其他代谢过程中发挥重要作用。希特林蛋白缺乏症有3种临床表型:citrin缺乏所致新生儿肝内胆汗淤积症(neonatal intrahepatic cholestasis caused by citrin deficiency,NICCD),成年发作Ⅱ型瓜氨酸血症(adult onset type Ⅱ citrullinemia,CTLN2)以及介于两者之间的citrin缺乏所致发育异常及血脂异常(failure to thrive and dyslipidemia caused by citrin deficiency,FTTDCD)[1]。
NICCD患者通常在出生后1岁内发病,临床以肝内胆汗淤积、肝功能及凝血功能异常、低血糖、脂肪肝和血氨基酸浓度异常为主要表现。患者经过饮食治疗和密切随访其临床症状大部分可得以缓解,少数患者会发展为肝衰竭或者肝硬化,此时肝移植成为了唯一且有效的治疗方案[2-3]。本文介绍1例NICCD患儿接受杂合子亲体肝移植治疗的病例。
1 资料与方法
1例8个月的患儿通过基因检测及血清串联质谱分析结果诊断为NICCD。患儿生后1个月起出现反复黄疸伴高瓜氨酸血症、高氨血症和双羧酸尿,予以合理治疗后氨基酸代谢恢复正常,但肝功能持续恶化并发展为终末期肝病,在本院接受杂合子母亲供肝的亲体肝移植术。
2 结 果
患儿,男,2017年8月6日出生(孕37周)。患儿出生后1个月出现无明显诱因的黄疸,伴有喂养困难,轻度肝酶升高,凝血功能异常,无陶土便,予以保肝退黄疗效不佳。出生后2个月行基因检测提示SLC25A13复合杂合子突变(c. 852_855del TATG,c.775C>T),串联质谱分析提示血浆瓜氨酸(Cit 330.34 μmol/L)、精氨酸(Arg 111.7 μmol/L)、酪氨酸(Tyr 394.8 μmol/L)、甲硫氨酸(Met 107.1 μmol/L)和苏氨酸(Thr 182.8 μmol/L)均明显升高,尿液串联质谱提示双羧酸尿,肝酶升高,肝内胆汗淤积(图1A)和高氨血症(图1B),诊断为NICCD型希特林蛋白缺乏症。予以限制饮食(无乳糖),加强中链甘油三脂营养方案,补充脂溶性维生素及维生素K1,熊去氧胆酸退黄,苯巴比妥钠预防高氨血症等对症处理,患儿接受治疗2个月后复查提示血浆氨基酸基本恢复正常(Cit 29.1 μmol/L,Arg 24.2 μmol/L,Tyr 84.8 μmol/L,Met 61.8 μmol/L,Thr 91 μmol/L),双羧酸尿症消失,高血氨症明显改善(图1B)。然而患儿黄疸持续升高(图1A),伴有腹水、腹壁静脉曲张等肝硬化失代偿表现。患儿出生后7个月行腹腔镜探查术提示存在胆道闭锁。出生后8个月到上海交通大学医学院附属仁济医院就诊拟行肝移植术。术前评估提示患儿存在肝硬化失代偿合并脾亢,并伴有凝血功能异常(凝血酶原时间为22秒,国际标准化比值为1.99),Child-pugh C级,发育正常(身长67 cm,Z=-1.0,体重8.3 kg,Z=-1.3)。患儿父母基因型检查均为SLC25A13突变基因携带者,但无CTLN2表现,肝功能均正常。
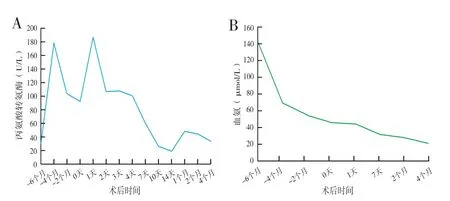
图1 患儿术前术后血清丙氨酸转氨酶变化(A);患儿术前术后血氨变化(B)
患儿于2018年4月19日行同血型亲体肝移植术,供体为患儿母亲,切取左外叶作为供肝,质量180 g(移植受体的体重比为1.7 %),手术顺利,术后予以激素+他克莫司的二联抗免疫排斥方案。患儿术后第4 d从重症监护病房(intensive care unit,ICU)转入普通病房,术后第18 d出院。术后1周患儿肝功能及黄疸下降到正常水平(图2),凝血功能恢复正常(凝血酶原时间为14 s,国际标准化比值为1.19),他克莫司血药浓度维持在正常水平,并在此后的随访中维持稳定。病肝病理提示胆道闭锁样表现,伴轻度脂肪肝。术后串联质谱分析提示血浆氨基酸浓度均恢复正常,患儿目前无饮食限制,生长发育正常(身长80 cm,Z=-1.0,体重 9.5 kg,Z=-0.5)。患儿术后第6 d出现黏连性肠梗阻,予以禁食、胃肠减压和肠胃营养等支持治疗,肠梗阻缓解,随访至今无其他并发症。供体术后恢复顺利,术后第5 d出院,并在随访期间无并发症发生。
3 讨 论
希特林蛋白是主要存在于肝脏中的一种天冬氨酸/谷氨酸载体蛋白,其主要功能为将线粒体中的天冬氨酸转运到细胞质中与瓜氨酸合成为精氨琥珀酸。因此,希特林蛋白的功能异常可以导致尿素循环过程中精氨琥珀酸合成障碍,从而导致患者出现高瓜氨酸血症和高氨血症[4]。
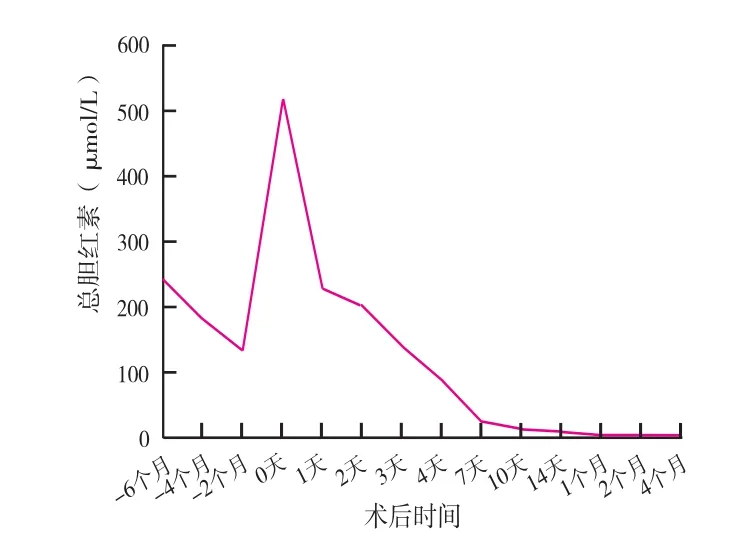
图2 患儿术前术后血清总胆红素的变化
NICCD主要在患儿出生后几个月内发病,患儿出生时常伴随低体重或者发育障碍,随后出现肝内胆汗淤积、肝大、弥漫性脂肪肝、肝功能异常、低蛋白血症、凝血功能异常、溶血性贫血及低血糖血症等表现。这类患儿通过适当的治疗,如补充脂溶性维生素、限制乳糖摄入和加强中链甘油三酯饮食,绝大数患儿的临床症状在1岁后会得以改善。部分患儿在10~20岁进展为CTLN2[5-6]。少数患儿饮食治疗效果不佳,甚至发展为肝硬化和肝衰竭[2]。本例患儿在出生后2个月确诊为NICCD,予以适当的饮食治疗方案,患儿氨基酸代谢基本恢复正常,高瓜氨酸血症及其他异常氨基酸代谢都得以控制,双羧酸尿症缓解,但是患儿黄疸持续增高,肝功能及凝血功能持续恶化,逐渐发展为终末期肝病,肝移植成为唯一有效的治疗手段。肝移植术后患儿黄疸消失,肝功能及凝血功能均恢复正常并在随访期间保持稳定,患儿术后无需特殊饮食控制,氨基酸代谢保持正常,患儿生活质量得到了明显提高。
由于NICCD是常染色隐性遗传病,因此患儿的父母均为突变基因的携带者。由于通常正常人体仅需30%左右正常活性的希特林蛋白即可维持正常的天冬氨酸代谢,因此大部分突变基因携带者无临床表现[7-8]。本文病例中患儿父母均为SLC25A13突变基因携带者,但术前评估提示肝功能及氨基酸代谢均正常。患儿母亲左外叶作为供肝移植到患儿体内后,患儿黄疸及肝功能均恢复正常,术后开始正常饮食,其氨基酸代谢仍保持正常。因此,对于肝功能及氨基酸代谢正常的SLC25A13突变基因携带者可以作为亲体肝移植供体,纠正NICCD受体原有的代谢异常。
NICCD患儿通常单独发病,也可合并其他代谢性疾病。Tamamori等[3]报道NICCD合并酪氨酸血症的患儿接受肝移植治疗的疗效。本例患儿术前行腹腔镜探查提示胆道闭锁,术后病肝病理再次确诊NICCD合并胆道闭锁的诊断。NICCD患儿本身容易在发病初期出现黄疸及肝功能异常,但是多经过相应的治疗得以改善。本例患儿经过恰当的内科治疗后虽然氨基酸及三羧酸循环(TCA)代谢恢复正常,但是黄疸及肝功能异常持续存在,并发展为终末期肝病。NICCD少数患儿经过恰当治疗仍会发展为肝衰竭,但是既往未见NICCD合并胆道闭锁的报道。对于经过治疗后肝功能持续恶化的NICCD患儿,需要考虑合并其他先天性或代谢性肝病,并予以相应的治疗。
总之,对于合并终末期肝病的NICCD患儿,肝移植可以有效的治疗患儿的肝功能异常及既有的代谢性疾病。对于经过积极治疗仍存在严重肝功能异常的NICCD患儿,需要考虑是否合并其他先天性胆汗淤积性肝病。
