中温固体氧化物燃料电池电解质研究进展
2019-01-25孙海滨张振昊张玉军
孙海滨,郭 学,张振昊,张玉军
中温固体氧化物燃料电池电解质研究进展
孙海滨1,郭 学1,张振昊2,张玉军3
1山东理工大学 材料科学与工程学院,山东 淄博 2550492莱芜亚赛陶瓷技术有限公司,山东 莱芜 2711003山东大学 材料科学与工程学院,济南 256216
固体氧化物燃料电池 (SOFC) 是一种新型能源转换装置,电解质是其核心部件之一。SOFC的中温化发展要求电解质材料在中温条件下 (500°C ~ 700°C) 具有较高离子电导率,在电池运行环境下具有良好的长期稳定性。本文主要综述了氧离子传导型电解质、质子传导型电解质和复合电解质的研究进展,分析了制约其发展与应用的难题,指出了中温电解质材料的发展方向。
固体氧化物燃料电池;氧离子型电解质;质子型电解质;复合电解质
固体氧化物燃料电池 (Solid Oxide Fuel Cell, SOFC) 是一种新型的能源转换装置,可直接将化学能转换为电能,因具有能量转化率高、无污染以及燃料多样性等优点而成为近年来的研究热点[1]。SOFC由阳极、阴极和电解质3部分组成。电解质是其中的核心部件,其作用是传导离子以及隔绝燃料气和氧化气。传统的SOFC多采用氧化钇稳定氧化锆作为电解质,但其所需的工作温度较高 (³1000°C),导致电池高温密封困难、制造成本高和长期运行稳定性差。研究和开发中温 (500°C ~ 700°C)固体氧化物燃料电池可以扩大电池组件材料的选择范围并且有效地提高电池长期运行的稳定性,是目前SOFC的主要发展方向之一[2]。然而,工作温度的降低会使电解质的欧姆阻抗迅速增大,导致电池性能急剧下降。因此,亟待开发新型的中温电解质材料。
根据载流子传导类型不同,SOFC电解质可分为氧离子传导型电解质、质子传导型电解质和复合电解质三大类。本文主要介绍这三类电解质的性能特点、发展现状以及提高电导率的主要途径,并对其未来发展做了展望。
1 氧离子传导型电解质
目前比较有代表性的氧离子型电解质主要包括萤石结构的ZrO2基电解质、CeO2基电解质、Bi2O3基电解质和钙钛矿结构的LaGaO3基电解质。
1.1 ZrO2基电解质
ZrO2在高温下为典型的立方萤石型结构。但是,纯ZrO2的离子电导率非常低。这是因为氧化锆在常温下为单斜晶型,只有在2300°C以上才能转变为立方萤石型结构[3]。研究表明[4,5],低价元素掺杂可以在室温至熔点范围内将ZrO2稳定在立方结构;同时,掺杂还会使晶格内会产生大量氧空位,从而使ZrO2成为良好的氧离子导体。氧空位的形成可以表示为:

(1) (2)
式中,M代表二价阳离子,R代表三价阳离子,ZrZrx、OOx、MZr²(或RZr¢)、VO··分别是位于正常晶格结点的Zr4+离子、位于正常晶格结点的O2-离子、Zr4+固有晶格结点上的M2+(或R3+)离子以及氧空位。
比较有代表性的低价掺杂离子是Ca2+和Y3+,可分别形成氧化钙稳定氧化锆 (CaO-Stabilized Zirconia, CSZ) 和氧化钇稳定氧化锆 (Yttria-Stabilized Zirconia, YSZ),其中后者在800°C以上具有较高的氧离子电导率[6-8]。同时,在氧化和还原气氛下,YSZ电解质都表现出了高的稳定性、强度和韧性。因此,YSZ电解质在高温 (800°C ~ 1000°C) SOFC中得到了广泛应用。其不足之处是在800°C以下电导率很低,因此YSZ并不适用于SOFC的中低温运行。近年来,Sc掺杂ZrO2(ScSZ) 被报道具有更高的氧离子电导率和更低的电导活化能[9-11]。不过,ScSZ的价格过高,制约了其在SOFC领域的实际应用。
1.2 CeO2基电解质
和ZrO2类似,纯CeO2材料的离子电导率也很低,一般需要通过低价态氧化物的掺杂提高其离子导电能力。目前萤石结构的掺杂CeO2(Doped Ceria,DCO) 已成为最常用的中低温SOFC电解质材料[12-15]。典型的低价态掺杂氧化物是Gd2O3和Sm2O3,分别形成Gd掺杂CeO2(GDC) 和Sm掺杂CeO2(SDC)。图1给出的是几种氧离子电解质的电导率对比,可以看出,GDC具有比YSZ高几倍甚至到几个数量级的电导率。不过,DCO在低氧分压或者较高温度下 (³600°C) 会发生如下反应:

(3)
Figure 1 The electrical conductivities of YSZ, GDC and LSGM electrolytes[19,28,29]
一种避免Ce4+被还原的方法是在阳极和DCO电解质之间添加厚度为1 μm ~ 2 μm的电子阻隔层 (如YSZ),但多层电解质界面上的反应物会降低电解质的电导率,并且热膨胀系数不匹配容易造成电解质开裂[17-19]。也有人尝试采用稀土元素共掺杂的方式来提高DCO在还原气氛下的稳定性,结果表明[20,21],共掺杂Sm和Nd元素可以获得比10 mol% Gd-CeO2更高的电导率,但是其还原稳定性并无改进。
尽管暂时无法解决Ce4+易被还原的问题,在500°C ~ 700°C温度范围内,DCO电解质仍可应用于SOFC。这是因为在该温度区间内,DCO的氧离子迁移数可达到0.9,足以提供较高的电池性能[20,22]。如果将SOFC的工作温度降低到500°C以下,其氧离子迁移数将有望得到进一步提高,电子电导甚至可以忽略,不过这是以牺牲氧离子电导率为代价的。使DCO在中温条件下具有较高的氧离子电导率和可以忽略的电子电导,是当前的重点研究方向之一。
陶瓷电解质为多晶结构,其微观结构 (尤其是晶界和晶粒) 对电导率的影响非常显著。为了进一步提高CeO2基电解质在中低温的离子电导率,晶界和晶粒尺寸对电学性能的影响得到了广泛关注。一般情况下,CeO2基电解质的晶界对氧离子传输起阻碍作用,其晶界电阻通常比晶粒电阻高几倍。其原因在于,因为原料纯度或炉膛材料高温挥发等原因,在电解质制备过程中会引入一部分Si杂质,这些Si杂质富集在电解质的晶界处,阻碍离子传输[23]。如果使用高纯度原料则可以有效降低杂质效应对晶界离子传输的影响。在这种情况下,晶界的电学性能主要受空间电荷效应的影响。
在空间电荷模型中 [图2 (a)],晶界是由一个晶界核 (Grain-Boundary Core] 和两个相邻的空间电荷层 (Space-Charge Layer) 组成的[24,25]。大量的氧空位富集在晶界核处,所以晶界核的电势为正[26]。在静电作用下,空间电荷层的氧空位数量急剧降低 [图2 (b)],使得该区域具有电抗性,从而阻碍离子的传输。
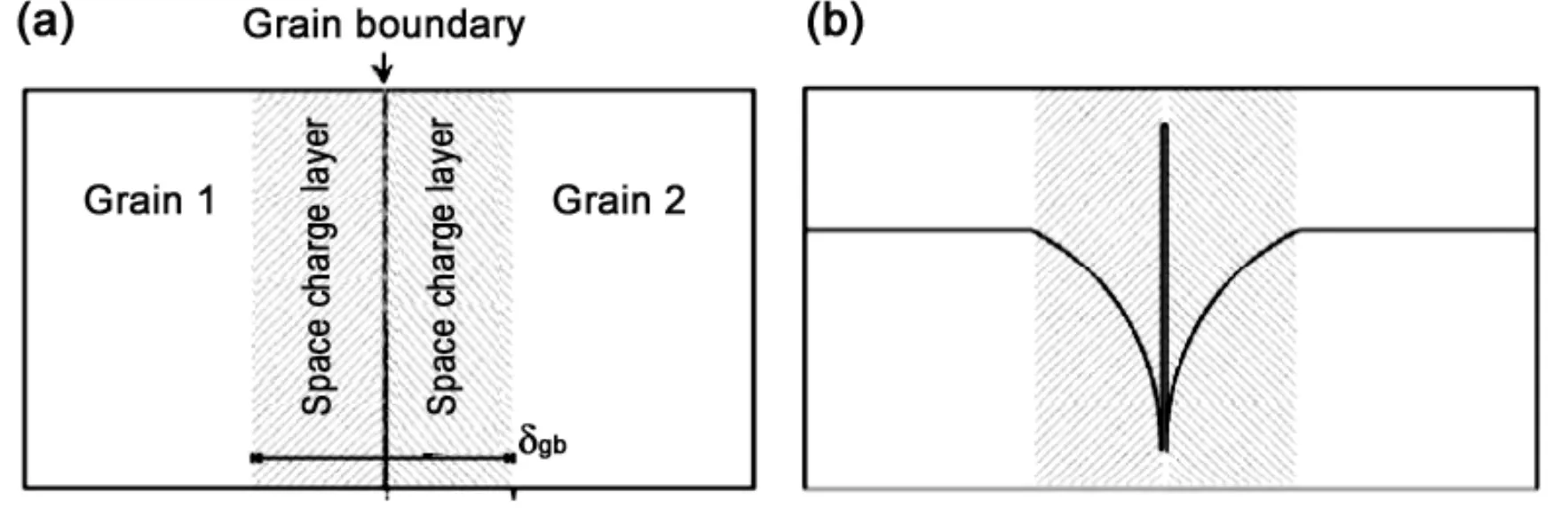
图2 (a) 晶界的空间电荷层示意图以及 (b) 氧空位在空间电荷层的分布
Figure 2 Schematic representations of (a) “electrical grain boundary” and (b) oxygen vacancy profiles in the space-charge layer model
降低晶界电阻的方法有两种:(1) 降低烧成温度;(2) 晶粒尺寸纳米化。
Singh等人[27]的研究表明,随着烧成温度的降低,电解质的晶界活化能由1.03 eV降至0.80 eV,经低温煅烧制备的、晶粒尺寸为0.21 μm ~ 0.89 μm的电解质晶界数量较多,可稀释晶界处的杂质,从而促进晶界处的氧离子传输。
降低电解质烧成温度的主要方法有两种:(1) 添加烧结助剂 (如CoO、CuO、MnO2、Fe2O3、Bi2O3、Cr2O3、NiO等); (2) 采用高活性纳米粉体作为原料。其中,添加过渡金属作为烧结助剂,不仅能够降低CeO2基电解质的烧成温度,还能够净化晶界和消除空间电荷效应[28]。这是因为过渡金属的离子半径小于Ce4+,且固溶度范围窄,容易沿晶界聚集并取代Ce4+,从而引入负电荷。负电荷可以降低晶界核的正电势,减弱空间电荷层氧空位所受到的排斥力,从而提高空间电荷层的氧空位浓度,降低晶界电阻。CeO2基电解质中过渡金属的添加量一般在0.5 mol% ~ 1 mol%。在众多过渡金属中,Fe3+和Co2+在清除杂质和降低空间电荷效应方面的作用尤为突出。
如前文所述,多晶电解质的晶界对离子传输起阻碍作用。不过在纳米结构 (或超细晶粒) 电解质中,晶界在离子传输过程中占主导,电解质的电导率得到显著提高,这就是所谓的“纳米效应”。Tschope等人[29]研究表明,随着CeO2基电解质晶粒尺寸的减小,其电导率增大,活化能降低。Christie等人[30]的研究表明,随着Gd掺杂CeO2电解质晶粒尺寸的减小,晶界为离子传输提供了连续的快速通道,晶界电导率增大。在纳米结构电解质中,晶界的表面能高,有助于提高氧离子的迁移率。Martín等人[31]的研究表明,在纳米SDC电解质中,晶界处的氧空位跃迁机制表现为非定域跃迁,与微米结构中的定域跃迁相比更有利于氧离子的快速传输,高电导率的形成主要归因于氧离子的快速传输。
不过,另一种说法是纳米电解质的高电导率是因为引入了电子电导[32]。Chiang等人[33]也认为,纳米结构使得晶界处的缺陷形成能降低,从而产生电子电导。作者所在课题组采用两步烧结法制备了平均晶粒尺寸约为140 nm的CeO2-20 mol% Sm2O3电解质,其晶粒电导率约为微米结构 SDC 电解质的20倍[34]。通过设计浓差电池,测试电池在不同气氛下的开路电压和氧离子迁移数,结果表明,超细晶粒电解质和微米结构电解质具有相同的氧离子迁移数,说明晶界电导率的提高并不是由额外的电子电导引起的,而可能是由氧离子扩散速率加快造成的,这与Martín等人[31]的研究结果相一致。关于CeO2基纳米电解质晶界的载流子传导类型,仍需进一步深入研究。
1.3 Bi2O3基电解质
在目前已知的氧离子导体中,按照电导率高低排序:Bi2O3> CeO2> ZrO2,Bi2O3基电解质的电导率最高。
Bi2O3是一种多晶型氧化物,在室温下呈单斜结构 (Bi2O3),650°C以下呈体心立方结构 (Bi2O3) 和四方结构 (Bi2O3),730°C ~ 825°C呈立方萤石结构 (Bi2O3)。其中,Bi2O3电导率最高,在825°C (熔点) 附近的离子电导可高达0.1 S/cm,相同温度下比YSZ电解质高2个数量级[35]。尽管Bi2O3基电解质具有非常高的电导率,但在实际应用中却存在一些问题,如:相变产生非常大的体积变化,导致材料断裂和性能恶化;在低氧分压下非常不稳定,易被直接还原为金属Bi,导致电导率下降,并产生电子电导[36,37];热稳定性和相结构在电池测试条件下的长期运行稳定性差。
为了使Bi2O3能够在较低温度下获得稳定的Bi2O3相,通常采用元素掺杂的方式部分取代铋离子。常用的掺杂金属氧化物为Y2O3、Er2O3、SrO、CaO、BaO、WO3、Ln2O3、Gb2O3、Nb2O3或Sm2O3。其中,20 mol% Er2O3掺杂的Bi2O3获得的电导率最高 (500°C下为2.3 S/cm;700°C下为37 S/cm)[38],比YSZ高50 ~ 100倍。
为了解决Bi2O3电解质在低氧分压下不稳定的问题,可通过制备双层电解质或在Bi2O3表面包覆其它材料来避免其与燃料气体接触。Wang等人[39]制备了基于CeO2/Bi2O3双层电解质的SOFC,与Bi2O3单一电解质组成的SOFC相比,最大输出功率提高了约33%。何岚鹰等人[40]在Bi2O3表面包覆了YSZ保护膜,电解质在500°C ~ 800°C范围内表现出了较好的稳定性。
为了解决Bi2O3在高温下长期运行稳定性差的问题,Huang等人[41]向Bi2O3中加入ZrO2来抑制相变,当掺杂量为5% 时,Bi2O3电解质的结构稳定,在650°C运行1000 h,电导率无明显降低。Joh等人[42]将20 wt% Er掺杂Bi2O3(ESB) 和YSZ复合,电解质在750°C运行600 h,也表现出了非常好的稳定性 (图3)。
如何进一步提高Bi2O3基电解质材料低氧分压条件下的稳定性将是今后的重点研究方向之一。
1.4 LaGaO3基电解质
LaGaO3是典型的钙钛矿化合物 (ABO3),对其A位进行碱土金属元素掺杂或/和用低价碱土或过渡金属阳离子对B位掺杂均可使LaGaO3结构中产生氧离子空位,从而提高其离子电导率。LaGaO3的电导率与掺杂离子半径以及掺杂量有关。A位掺杂通常选择与La3+离子半径相近的Sr2+、Ca2+、Ba2+,B位掺杂则通常选择与Ga3+离子相近的Mg2+。其中,La1-xSrxGa1-yMgyO3-δ(LSGM) 被认为是电导率最高的双掺杂钙钛矿材料[43],其电导率与DCO相当。与DCO相比,LSGM在很宽的氧分压范围内 (10-20< PO2< 1) 为纯氧离子导体[44,45]。
不过,LSGM用于Ni基阳极支撑SOFC时,在阳极/电解质高温共烧过程中容易和Ni发生化学反应,导致电解质电导率下降[44,46-48]。Bi等人[49]和Lin等人[50]通过在LSGM和Ni基阳极之间添加La掺杂CeO2(LDC) 过渡层,避免了LSGM和Ni发生反应。不过,当LDC过渡层较薄时,无法完全避免Ni扩散到LSGM电解质;当LDC过渡层较厚时,又会导致电解质的欧姆阻抗过大。Bozza等人[51]首先采用高温共烧制备LDC多孔阳极/LSGM电解质双层结构,然后向LDC多孔结构中浸渍硝酸镍,经低温煅烧生成NiO纳米颗粒包覆的LDC阳极 (图4)。该方法既不需要LDC过渡层,又能避免Ni和LSGM的高温反应,获得了较高的电池性能 (700°C时达到750 mW/cm2)。
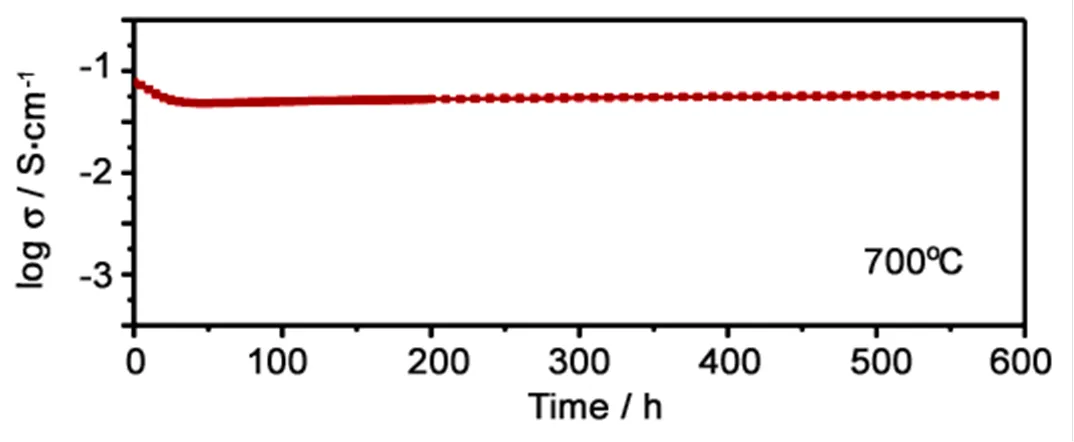
图3 20 wt% ESB-YSZ复合电解质在750°C空气测试条件下的电导率
Figure 3 The electrical conductivity of 20 wt% ESB-YSZ composite electrolytes measured at 750°C in air

图4 阳极支撑NiO-LDC/LSGM/LSCF固体氧化燃料电池的 (a) 断面和 (b) 阳极支撑体局部SEM照片
Figure 4 SEM images of (a) the cross-section view of anode supported NiO-LDC/LSGM/LSCF solid oxide fuel cell and (b) the high-magnification images of the anode support
因传统的阳极大多呈海绵状多孔结构,易导致溶液浸渍不充分。作者所在课题组采用冷冻流延成型技术制备了一种具有垂直孔道结构的阳极材料,经电解质溶液滴涂、电解质/阳极共烧、硝酸镍浸渍、低温煅烧制备了 (NiO-GDC)-LSGM阳极支撑的SOFC (图5)。这一独特的阳极独特结构有利于NiO浸渍,且避免了NiO和LSGM电解质之间的高温反应,从而获得了更高的电池性能 (700°C时达到1.61 W/cm2)[52]。

图5 冷冻流延法制备的 (NiO-GDC)-LSGM/LSGM/LSCF电池 (a) 断面及 (b) 阳极支撑体局部SEM照片
Figure 5 SEM images of (a) the cross-section view of (NiO-GDC)-LSGM/LSGM/LSCF solid oxide fuel cell fabricated by a freeze-tape-casting method and (b) the high-magnification image of the anode support
尽管LSGM的研究取得了一些进展,但其组分比较复杂,制备出符合化学计量比的LSGM较为困难,容易产生LaSrGa3O7、LaSrGaO4等杂质。此外,在高温煅烧过程中,Ga元素容易挥发。这些因素都会导致LSGM电导率降低,严重制约其实际应用[53]。
2 质子传导型电解质
质子传导型电解质主要有钙钛矿结构、复合钙钛矿结构等类型。
2.1 钙钛矿结构
ABO3型钙钛矿结构是最早发现的高温质子导体,也是当前的研究重点。在ABO3中,A位是二价离子,一般是Ba、Sr和Ca等碱土金属元素;B位是四价阳离子,一般是Ce、Zr等元素[54-61]。钙钛矿化合物本身并不能释放出质子成分,质子的来源是通过对B位进行三价元素掺杂后产生的氧缺陷与环境中的水分子或氢分子相互作用的结果。常见的掺杂元素是Y、Sm、Gd、Yb、Eu等三价稀土元素。
高温质子导体的质子化过程主要分为两种情况[62]。在干燥的氢气中,

(4)
在潮湿的气氛中[63],

(5)
此外,在高氧分压和高温条件下还可能发生以下反应:

(6) (7)
可见,这些钙钛矿化合物在不同气氛下会表现出不同的导电性:在干燥氢气中表现为质子和电子导体,在潮湿空气中是良好的质子导体;在高氧分压和高温条件下表现为氧离子和电子空穴的混合导体。
式 (5) 是形成质子缺陷的主要反应,该反应为放热反应[63]。质子浓度随着温度的降低而升高,在中低温下质子为主要载流子。相反地,氧离子浓度随着温度的升高而升高,在高温下氧离子为主要载流子[60,61]。为了获得良好的质子电导率,质子导体的工作温度不宜过高,一般低于700°C[64]。

图6 BaCe0.9-xZrxY0.1O3-δ在湿H2下的电导率
Figure 6 The electrical conductivity of BaCe0.9-xZrxY0.1O3-δelectrolytes measured in wet H2
目前研究最多的钙钛矿型质子导体是掺杂BaCeO3和掺杂BaZrO3。其中,掺杂BaCeO3因具有较高的电导率而被广泛地应用于SOFC中[55,65-68]。然而,掺杂BaCeO3基氧化物在CO2和H2O气氛中的化学稳定性很差[63,69,70],在10-4atm ~ 10-5atm的CO2下即可与CO2发生反应生成BaCO3和CeO2。在有水的气氛下,掺杂BaCeO3基氧化物还容易与水反应生成Ba(OH)2和CeO2。SOFC使用的燃料气体中一般都含有水分以提高质子电导,因此,单纯的掺杂BaCeO3体系无法应用于含水的燃料气体中。掺杂BaZrO3在CO2或H2O气氛中具有非常优异的化学稳定性[63,71],可以满足SOFC苛刻的工作气氛,但是其质子电导率相对较低,大约比掺杂BaCeO3要低一个数量级左右[72-74]。此外,BaZrO3的烧结活性差,需要在1700°C ~ 1800°C高温下才能烧结致密[59,64,75]。过高的烧结温度不仅会导致Ba挥发而引起材料组分的变化,降低电导率[76],还会导致阳极与电解质之间反应 (和/或相扩散) 以及阳极烧死等一系列问题。
因为BaZrO3和BaCeO3之间可以形成无限固溶体,所以向BaCeO3中掺杂Zr用以部分取代Ce可以有效地提高电解质的化学稳定性[64,74,77-80]。不同的Zr掺杂量对材料的化学稳定性有着显著的影响。当Zr掺杂量较低时,材料与CO2和H2O发生部分反应;当Zr掺杂量达到40% 时,材料在CO2和H2O气氛条件下显示出非常好的化学稳定性。不过,随着掺杂量的提高,材料本身的电导率会随之降低 (图6),同时,其烧结活性也会变差。因此,合适的Zr掺杂量应使得材料同时具有好的化学稳定性、高的电导率和烧结活性。目前,应用最多的一种质子电解质是BaZr0.1Ce0.7Y0.2O3-δ(BZCY)[66]。该材料在CO2气氛和水蒸气气氛下具有较高的稳定性,同时在600°C以下具有比YSZ以及GDC等氧离子导体更高的电导率 (图7),因而被认为是一种较理想的质子导体电解质。在此基础上,Yang等人[1]采用Y和Yb共掺杂的方法制备了BaZr0.1Ce0.7Y0.1Yb0.1O3-δ(BZCYYb),材料的电导率得到了进一步提高。不过,固相法合成的BZCYYb粉体的烧结活性差,需在1550°C煅烧10 h才能获得致密坯体,过高的烧成温度仍然会造成Ba挥发。
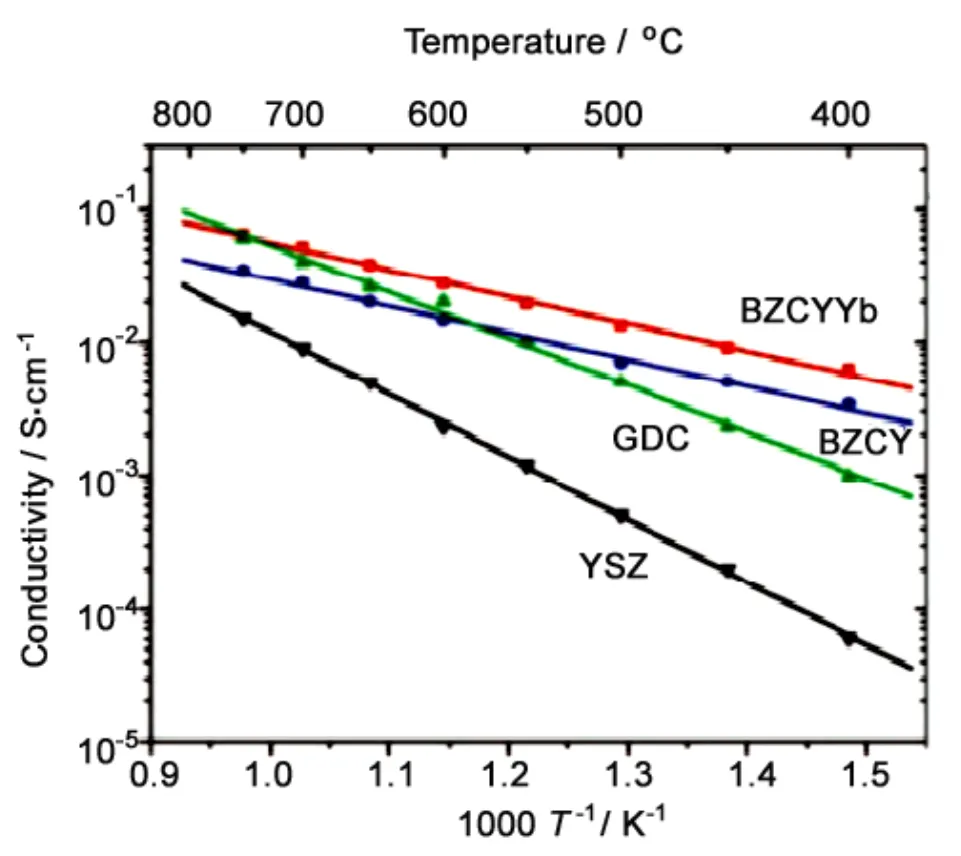
图7 不同材料在湿O2下的电导率
Figure 7 The electrical conductivities of different electrolyte materials measured in wet O2
尽管BZCY和BZCYYb材料的发展极大地推动了质子传导型SOFC的发展,但这种材料在高浓度的CO2气氛以及更苛刻的环境下并非十分稳定。因此,有必要进一步改善这些材料的烧结性能以及化学稳定性。
2.2 复合钙钛矿结构
复合钙钛矿结构的分子式为A3B¢1+xB²2-xO9-d,其中A表示Ba或者Sr,B¢和B²分别表示二价和五价金属离子。Ba3Ca1.18Nb1.82O9-d(BCN18) 因电导率高而成为这类化合物中研究最多的体系[81-86]。以BCN18为电解质的SOFC在CO/CO2环境下工作100多个小时仍然保持稳定,显示出其极好的化学稳定性[82]。然而,关于BCN18的电导率测试结果却有较大争议。Nowick等人[82]发现BCN18在600°C时的质子电导率约为10-2S·cm-1,离子迁移数大于0.98。Schober等人[84-86]的研究也表明该材料在700oC具有较高的电导率 (0.07 S·cm-1)。但是,Norby等人[87]的研究却发现相同条件下BCN18的电导率只有5.5 × 10-4S·cm-1。除了BCN18体系在电导率方面存在争议之外,其另一缺点就是该材料的烧结活性很差,通常需要在1600°C高温下才能烧结致密[88]。
3 复合电解质
复合电解质是由两种或多种离子电导性质不同的材料复合而成的两相或多相体系[89]。复合电解质可以使各电解质的性能实现优势互补。例如,DCO/碳酸盐复合电解质既可以抑制DCO在低氧分压或还原气氛下产生电子电导,又能够解决碳酸盐在高温运行环境中稳定性差的问题。DCO/BaCeO3基复合电解质除了抑制DCO产生的电子电导,还能够解决BaCeO3在含CO2、H2O气氛中化学性质不稳定的问题。
3.1 DCO-碳酸盐复合电解质
2001年,Zhu 等人[90]将GDC-碳酸盐用作SOFC的电解质材料,复合电解质在600°C时的电导率高达0.2 S/cm,在相同温度下电池最大功率密度达到580 mW/cm2。其电化学性能优于氧化物-硫酸盐/硝酸盐/磷酸盐等复合电解质体系。因此,DCO-碳酸盐复合电解质受到广泛关注。
在DCO-碳酸盐复合电解质中,DCO相通常为SDC、GDC等,碳酸盐已经由Na2CO3、K2CO3发展到了(Li/Na)2CO3、(Li/K)2CO3、 (Na/K)2CO3等二元共晶盐和(Li/Na/K)2CO3三元共晶盐。DCO相和碳酸盐相的界面存在相互作用,产生增强效应,可为离子传输提供快速通道。因此,提高复合电解质的两相界面数量有助于提高其电导率。Jing等人[91]采用SPS法制备了纳米结构的SDC-(Li0.5Na0.25K0.25)2CO3电解质,在600°C电导率高达1.23´10-1S/cm。Xia等人[92]的研究表明,只有当DCO和碳酸盐的比例在合理范围时,复合电解质的界面才能表现出显著增强效应。如果碳酸盐比例过低 (£10%),就无法在复合电解质中形成连续的两相界面;如果碳酸盐比例过高 (³50%),则碳酸盐易流失,导致电池稳定性差。
与单一DCO电解质的导电机理不同,DCO-碳酸盐复合电解质是一种H+、O2-共同传导体。Huang 等人[93]基于SDC-碳酸盐制备了SOFC,在氢气/空气测试条件下观察到电池的阴极和阳极两侧都有水生成,证实了DCO-碳酸盐复合电解质中同时存在O2-和H+传导。Zhao等人[94]用直流四电极法研究了该复合电解质的载流子传导行为,结果表明,DCO相和碳酸盐相中均存在O2-传导,H+传导仅存在于碳酸盐相和两相界面。还有研究发现[95],向阴极气体中加入CO2,基于DCO-碳酸盐复合电解质的SOFC可以获得更高的输出功率,证明了CO2的存在促进电解质的离子传导。
DCO-碳酸盐复合电解质中各种载流子的传导机理较为复杂,仍有待进行进一步探究。
3.2 DCO/BaCeO3基复合电解质
Venkatasubramanian等人[96]和Khandelwal等人[97]制备了DCO/BaCeO3基复合电解质。与单一DCO电解质相比,采用该复合电解质制备的SOFC表现出了较高的开路电压 (Open Circuit Voltage, OCV),说明电子电导得到了有效抑制。不过,该复合电解质的电导率低于单一DCO电解质和BaCeO3基电解质,其晶界对于离子传输起到了阻碍作用。其主要原因是复合电解质需要的烧成温度较高 (1450°C ~ 1550°C),在高温煅烧过程中易导致两相界面发生元素扩散和化学反应,杂质富集在晶界处,造成晶界电阻过大。Sun 等人[98]采用凝胶燃烧法合成了高活性SDC/BCS (BaCe0.8Sm0.2O3-δ) 纳米粉体,以此为原料经高温煅烧制备了复合电解质,烧成温度降低了100°C ~ 200°C,晶界扩散/化学反应得到抑制,晶界电阻得以降低。采用该复合电解质制备的SOFC,在700°C、氢气/空气测试条件下,最大输出功率达到550 mW/cm2。若选择合适的烧结助剂,能够在进一步降低烧成温度的基础上净化晶界杂质,将有望获得更高的电池性能。
4 结论与展望
随着固体氧化物燃料电池的中温化发展,开发新型的中温电解质材料成为一个研究热点。目前的氧离子传导型电解质和质子型电解质虽然在中温条件下表现出了各自的优势,但仍存在一些问题,如:ZrO2基电解质的中温电导率低、CeO2基电解质在低氧分压下易被部分还原而产生电子电导、Bi2O3基电解质极易被还原为金属铋、LaGaO3基电解质因成分复杂难以获得纯相材料等。钙钛矿和复合钙钛矿型质子传导型电解质则普遍存在烧结温度过高、化学稳定性差、电导率偏低的问题。DCO/碳酸盐、CeO2/BaCeO3基复合电解质可以解决单一电解质存在的问题,实现两相或多相材料性能的优势互补,但其导电机理更为复杂。
固体氧化物燃料电池电解质今后的研究方向为:(1) 复合化:利用两相或多相材料的性能优势,解决制约单一电解质在实际应用中的短板问题;(2) 纳米化:使晶界在离子传导过程中占主导,通过晶界的增强效应提高离子电导率;(3) 不同类型电解质的导电机理研究。
[1] YANG L, WANG S, BLINN K, et al. Enhanced sulfur and coking tolerance of a mixed ion conductor for SOFCs: BaZr0.1Ce0.7Y0.2-xYbxO3-δ[J]. Science, 2009, 326: 126-129.
[2] WACHSMAN ED, LEE KT. Lowering the temperature of solid oxide fuel cells [J]. Science, 2011, 334: 935-939.
[3] SCOTT HG. Phase relationships in the zirconia-yttria system [J]. Journal of Materials Science, 1975, 10: 1527-1535.
[4] POLITOVA T. Investigation of scandia-yttria-zirconia system as an electrolyte material for intermediate temperature fuel cells-influence of yttria content in system (Y2O3)x(Sc2O3)(11−x)(ZrO2)89[J]. Solid State Ionics, 2004, 168: 153-165.
[5] KITAZAWA K, COBLE RL. Use of stabilized ZrO2to measure O2permeation [J]. Journal of the American Ceramic Society, 1974, 57: 360-363.
[6] HAILE SM. Fuel cell materials and components [J]. Acta Materialia, 2003, 51: 5981-6000.
[7] GOODENOUGH JB. Oxide-Ion Electrolytes [J]. Annual Review of Materials Research, 2003, 33: 91-128.
[8] BRETT DJ, ATKINSON A, BRANDON NP, et al. Intermediate temperature solid oxide fuel cells [J]. Chemical Society Reviews, 2008, 37: 1568-1578.
[9] DEVANATHAN R, THEVUTHASAN S, GALE JD. Defect interactions and ionic transport in scandia stabilized zirconia [J]. Physical Chemistry Chemical Physics, 2009, 11: 5506-5511.
[10] SARAT S, SAMMES N, SMIMOVA A. Bismuth oxide doped scandia-stabilized zirconia electrolyte for the intermediate temperature solid oxide fuel cells [J]. Journal of Power Sources, 2006, 160: 892-896.
[11] BADWAL SPS, CIACCHI FT, MILOSEVIC D. Scandia-zirconia electrolytes for intermediate temperature solid oxide fuel cell operation [J]. Solid State Ionics, 2000, 136-137: 91-99.
[12] MOGENSEN M, LYBYE D, BONANOS N, et al. Factors controlling the oxide ion conductivity of fluorite and perovskite structured oxides [J]. Solid State Ionics, 2004, 174: 279-286.
[13] SAHIBZADAB M, STEELEA BCH, ZHENG K, et al. Development of solid oxide fuel cells based on a Ce(Gd)O2-xelectrolyte film for intermediate temperature operation [J]. Catalysis Today, 1997, 38: 459-466.
[14] STEELE BCH. Appraisal of Ce1-yGdyO2-y/2electrolytes for IT-SOFC operation at 500oC [J]. Solid State Ionics, 2000, 129: 95-110.
[15] ZHENG K, STEELE BCH, SAHIBZADA M, et al. Solid oxide fuel cells based on Ce(Gd)O2-xelectrolytes [J]. Solid State Ionics, 1996, 86-88: 1241-1244.
[16] WANG S, INABA H, TAGAWA H, et al. Nonstoichiometry of Ce0.9Gd0.1O1.95-x[J]. Solid State Ionics, 1998, 107: 73-79.
[17] TSOGA A, GUPTA A, NAOUMIDIS A, et al. Gadolinia-doped ceria and yttria stabilized zirconia interfaces: regarding their application for SOFC technology [J]. Acta Materialia, 2000, 48: 4709-4714.
[18] HORITA T, SAKAI N, YOKOKAWA H. Ceria-zirconia composite electrolyte for solid oxide fuel cells [J]. Journal of Electroceramics, 1997, 1: 155-164.
[19] TOMPSETT GA, SAMMES NM, YAMAMOTO O. Ceria-yttria-stabilized zirconia composite ceramic systems for applications as low-temperature electrolytes [J]. Journal of the American Ceramic Society, 1997, 80: 3181-3186.
[20] OMAR S, WACHSMAN E, NINO J. Higher conductivity Sm3+and Nd3+co-doped ceria-based electrolyte materials [J]. Solid State Ionics, 2008, 178: 1890-1897.
[21] KHARTON VV, FIGUEIREDO FM, NAVARRO L, et al. Ceria-based materials for solid oxide fuel cells [J]. Journal of Materials Science, 2001, 36: 1105-1117.
[22] MILLIKEN C, GURUSWAMY S, KHANDKAR A. Properties and performance of cation-doped ceria electrolyte materials in solid oxide fuel cell applications [J]. Journal of The American Ceramics Society, 2002, 85: 2479-2486.
[23] KOSACKI I, SUZUKI T, PETROVSKY V, et al. Electrical conductivity of nanocrystalline ceria and zirconia thin film [J]. Solid State Ionics, 2000,136-137:1225-1233.
[24] GUO X, WASER R. Electrical properties of the grain boundaries of oxygen ion conductors: acceptor- doped zirconia and ceria [J]. Progress in Materials Science, 2006, 51: 151-210.
[25] KLIE RF, BROWNING ND. Atomic scale characterization of oxygen vacancy segregation at SrTiO3grain boundaries [J]. Applied Physics Letters, 2000, 77: 3737-3739.
[26] HAILE SM, STANEFF G, RYU KH. Non-stoichiometry, grain boundary transport and chemical stability of proton conducting perovskites [J]. Journal of Materials Science, 2001, 36: 1149-1160.
[27] SINGH V, BABU S, KARAKOTI AS, et al. Effect of submicron grains on ionic conductivity of nanocrystalline doped ceria [J]. Journal of Nanoscience and Nanotechnology, 2010, 10: 6495-6503.
[28] ARUNKUMAR P, MEENA M, BABU KS. A review on cerium oxide-based electrolytes for ITSOFC [J]. Nanomaterials and Energy, 2012, 1: 288-305.
[29] TSCHOPE A, SOMMER E, BIRRINGER R. Grain size-dependence of electrical conductivity in polycrystalline cerium oxide: I, Experiments [J]. Journal of Electroceramics, 2001, 7: 169-177.
[30] CHRISTIE GM, VAN BERKEL FPF. Microstructure-ionic conductivity relationships in ceria-gadolinia electrolytes [J]. Solid State Ionics, 1996, 83: 17-27.
[31] BELLINO MG, LAMAS DG, WALSOE DE, et al. A mechanism for the fast ionic transport in nanostructured oxide-ion solid electrolytes [J]. Advanced Materials, 2006, 18: 3005-3009.
[32] HUI S, ROLLER J, YICK S, et al. A brief review of the ionic conductivity enhancement for selected oxide electrolytes [J]. Journal of Power Sources, 2007, 172: 493-502.
[33] CHIANG YM, LAVIK EB, KOSACKI HL, et al. Nonstoichiometry and electrical conductivity of nanocrystalline CeO2-x[J]. Journal of Electroceramics, 1997, 1: 7-14.
[34] SUN H, RAINWATER BH, XIONG X, et al. Interfacial effects on electrical conductivity in ultrafine-grained Sm0.2Ce0.8O2-δelectrolytes fabricated by a two-step sintering process [J]. International Journal of Hydrogen Energy, 2017, 42: 11823-11829.
[35] 苏莎, 陈海清, 谭令. 氧化铋基固体氧化物燃料电池电解质研究进展[J]. 湖南有色金属, 2014, 30 (4): 45-48.
[36] SHUK P, WIEMHOFERB HD, GUTH U, et al. Oxide ion conducting solid electrolytes based on Bi2O3[J]. Solid State Ionics, 1996, 89: 179-196.
[37] AZAD AM, LAROSE S, AKBARSA. Bismuth oxide-based solid electrolytes for fuel cells [J]. Journal of Materials Science, 1994, 29: 4135-4151.
[38] 魏丽陈, 王琴. 中温固体氧化物燃料电池电解质材料的研究进展[J]. 稀有金属, 2003, 27: 286-298.
[39] WANG FY, CHEN S, WANG Q, et al. Study on Gd and Mg co-doped ceria electrolyte for intermediate temperature solid oxide fuel cells [J]. Catalysis Today, 2004, 97 (2-3): 189-194.
[40] 苏文辉, 何陈, 刘江. 有保护膜的Bi2O3基稀土固体电解质[J]. 吉林大学自然科学学报, 1994, 4: 63-66.
[41] HUANG K, WANG C, XU X. Activity of Bi2O3in Bi2O3-Y2O3oxygen ion conductor [J]. Journal of Solid State Chemistry, 1992, 98 (1): 206-209.
[42] JOH DW, PARK JH, KIM DY, et al. High performance zirconia-bismuth oxide nanocomposite electrolytes for lower temperature solid oxide fuel cells [J]. Journal of Power Sources, 2016, 320: 267-273.
[43] ISHIHARA T, MATSUDA H, TAKITA Y. Doped LaGaO3perovskite type oxide as a new oxide ionic conductor [J]. Journal of the American Ceramic Society, 1994, 116: 3801-3803.
[44] HUANG K, TICHY R, GOODENOUGH JB, et al. Superior perovskite oxide-ion conductor; strontium- and magnesium-doped LaGaO3: III, performance tests of single ceramic fuel cells [J]. Journal of the American Ceramic Society, 1998, 81: 2581-2585.
[45] TAO S, IRVINE JTS, KILNER JA. An effective solid oxide fuel cell based upon single-phase pevroskites [J]. Advanced Materials, 2005, 17: 1734-1737.
[46] YAMAJI K, HORITA T, ISHIKAWA M, et al. Chemical stability of the La0.9Sr0.1Ga0.8Mg0.2O2.85electrolyte in a reducing atmosphere [J]. Solid State Ionics, 1999, 121: 217-224.
[47] ZHANG X, OHARA S, MARIC R, et al. Interface reactions in the NiO-SDC-LSGM system [J]. Solid State Ionics, 2000, 133: 153-160.
[48] HUANG K, FENG M, GOODENOUGH JB, et al. Electrode performance test on single ceramic fuel cells using as electrolyte Sr- and Mg-doped LaGaO3[J]. Journal of the Electrochemical Society, 1997, 144: 3620-3624.
[49] BI Z, YI B, WANG Z, et al. A High-performance anode-supported SOFC with LDC-LSGM bilayer electrolytes [J]. Electrochemical and Solid-State Letters, 2004,7 (5): A105-A107.
[50] LIN Y, BARMETT SA. Co-Firing of anode-supported SOFCs with thin La0.9Sr0.1Ga0.8Mg0.2O3−δelectrolytes [J]. Electrochemical and Solid-State Letters, 2006, 9 (6):A285-A288.
[51] BOZZA F, POLINI R, TRAVERSA E. High performance anode-supported intermediate temperature solid oxide fuel cells (IT-SOFCs) with La0.8Sr0.2Ga0.8Mg0.2O3-δelectrolyte films prepared by electrophoretic deposition [J]. Electrochemistry Communications, 2009, 11 (8): 1680-1683.
[52] SUN H, CHEN Y, CHEN F, et al. High-performance solid oxide fuel cells based on a thin La0.8Sr0.2Ga0.8Mg0.2O3-δelectrolyte membrane supported by a nickel-based anode of unique architecture [J]. Journal of Power Sources, 2016, 301: 199-203.
[53] MATRASZEK A. Phase diagram study in the La2O3-Ga2O3-MgO-SrO system in air [J]. Solid State Ionics, 2004, 166 (3-4): 343-350.
[54] DING H, LIN B, FANG D, et al. BaZr0.1Ce0.7Y0.2O3-δproton-conducting electrolyte prepared by gel-casting for low-temperature solid oxide fuel cells [J]. Journal of Alloys and Compounds, 2009, 474 (1-2): 364-369.
[55] PENG R, WU Y, YANG L, et al. Electrochemical properties of intermediate-temperature SOFCs based on proton conducting Sm-doped BaCeO3electrolyte thin film [J]. Solid State Ionics, 2006, 177 (3-4): 389-393.
[56] SCHOBER T. Transformation of an oxygen ion conductor to a proton conductor by solid state reaction [J]. Solid State Ionics, 2005,176 (29-30): 2275-2277.
[57] PELLETIER L, MCFARLAN A, MAFFEI N. Ammonia fuel cell using doped barium cerate proton conducting solid electrolytes [J]. Journal of Power Sources, 2005, 145 (2): 262-265.
[58] IWAHARA H. Oxide-ionic and protonic conductors based on perovskite-type oxides and their possible applications [J]. Solid State Ionics, 1992, 52: 99-104.
[59] SCHOBER T, BOHN HG. Water vapor solubility and electrochemical characterization of the high temperature proton conductor BaZr0.9Y0.1O2.95[J]. Solid State Ionics, 2000, 127: 351-360.
[60] YAJIMA T, IWAHARA H. Protonic and oxide ionic conduction in BaCeO3-based ceramics-effect of partial substitution for Ba in BaCe0.9Nd0.1O3-αwith Ca [J]. Solid State Ionics, 1991, 47: 117-124.
[61] IWAHARA H, YAJIMA T, USHIDA H. Effect of ionic radii of dopants on mixed ionic conduction (H++O2-) in BaCeO3-based electrolytes [J]. Solid State Ionics, 1994, 70-71: 267-271.
[62] WANG S, ZHANG L, ZHANG L, et al. Two-step sintering of ultrafine-grained barium cerate proton conducting ceramics [J]. Electrochimica Acta, 2013, 87: 194-200.
[63] KREUER KD. Proton-conducting oxides [J]. Annual Review of Materials Research, 2003, 33 (1): 333-359.
[64] KATAHIRA K, KOHCHI Y, SHIMURA T, et al. Protonic conduction in Zr-substituted BaCeO3[J]. Solid State Ionics, 2000, 138: 91-98.
[65] YANG L, ZUO C, WANG S, et al. A novel composite cathode for low-temperature SOFCs based on oxide proton conductors [J]. Advanced Materials, 2008, 20 (17): 3280-3283.
[66] ZUO C, ZHA S, LIU M, et al. Ba(Zr0.1Ce0.7Y0.2)O3-δas an electrolyte for low-temperature solid-oxide fuel cells [J]. Advanced Materials, 2006, 18 (24): 3318-3320.
[67] FANG S, BI L, WU X, et al. Chemical stability and hydrogen permeation performance of Ni- BaZr0.1Ce0.7Y0.2O3-δin an H2S-containing atmosphere [J]. Journal of Power Sources, 2008, 183 (1): 126-132.
[68] FANG S, BI L, YANG C, et al. H2S poisoning and regeneration of Ni–BaZr0.1Ce0.7Y0.2O3-δat intermediate temperature [J]. Journal of Alloys and Compounds, 2009, 475 (1-2): 935-939.
[69] GUO Y, LIN Y, RAN R, et al. Zirconium doping effect on the performance of proton-conducting BaZryCe0.8−yY0.2O3−δ(0.0 ≤ y ≤ 0.8) for fuel cell applications [J]. Journal of Power Sources, 2009, 193 (2): 400-407.
[70] CHEN F, SORENSEN OT, MENGA G, et al. Chemical stability study of BaCe0.9Nd0.1O3-αhigh- temperature proton-conducting ceramic [J]. Journal of Materials Chemistry, 1997, 7 (3): 481-485.
[71] TANIGUCHI N, NISHIMURA C, KATO J. Endurance against moisture for protonic conductors of perovskite-type ceramics and preparation of practical conductors [J]. Solid State Ionics, 2001, 145: 349-355.
[72] PHAIR JW, BADWAL SPS. Review of proton conductors for hydrogen separation [J]. Ionics, 2006, 12 (2): 103-115.
[73] SERRA JM, MRULENBERG WA. Thin-film proton BaZr0.85Y0.15O3conducting electrolytes: toward an intermediate-temperature solid oxide fuel cell alternative [J]. Journal of the American Ceramic Society, 2007, 90 (7): 2082-2089.
[74] FABBRI E, PERGOLESI D, D'EPIFANION A, et al. Design and fabrication of a chemically-stable proton conductor bilayer electrolyte for intermediate temperature solid oxide fuel cells (IT-SOFCs) [J]. Energy & Environmental Science, 2008, 1 (3): 355-359.
[75] KREUER KD, ADAMS S, MUNCH W, et al. Proton conducting alkaline earth zirconates and titanates for high drain electrochemical applications [J]. Solid State Ionics, 2001, 145: 295-306.
[76] YAMAZAKI Y, HEMANDEZ-SANCHEZ R, HAILE SM. Cation non-stoichiometry in yttrium-doped barium zirconate: phase behavior, microstructure, and proton conductivity [J]. Journal of Materials Chemistry, 2010, 20 (37): 8158-8166.
[77] ZUO C, DORRIS SE, BALACHANDRAN U, et al. Effect of Zr-doping on the chemical stability and hydrogen permeation of the Ni-BaCe0.8Y0.2O3-αmixed protonic-electronic conductor [J]. Chemistry of Materials, 2006, 18: 4647-4650.
[78] ZHONG Z. Stability and conductivity study of the BaCe0.9−xZrxY0.1O2.95systems [J]. Solid State Ionics, 2007, 178 (3-4): 213-220.
[79] BARISON S, BATTAGLIARIN M, CAVALLIN T, et al. High conductivity and chemical stability of BaCe1−x−yZrxYyO3−δproton conductors prepared by a sol-gel method [J]. Journal of Materials Chemistry, 2008, 18 (42): 5120-5128.
[80] BARISON S, BATTAGLIARIN M, CAVALLIN T, et al. Barium non-stoichiometry role on the properties of Ba1+xCe0.65Zr0.20Y0.15O3-δproton conductors for IT-SOFCs [J]. Fuel Cells, 2008, 8 (5): 360-368.
[81] DEPIFANIO A, FABBRI E, DIBARTOLOMEO E, et al. Design of BaZr0.8Y0.2O3–δprotonic conductor to improve the electrochemical performance in intermediate temperature solid oxide fuel cells (IT-SOFCs) [J]. Fuel Cells, 2008, 8 (1): 69-76.
[82] DU Y, NOWICK AS. Galvanic cell measurements on a fast proton perovskite electrolyte [J]. Solid State Ionics, 1996, 91: 85-91.
[83] GROß B, MARION S, LIND K, et al. Proton conducting Ba3Ca1.18Nb1.82O8.73-H2O pressure-compositions isotherms in terms of fermi-dirac statistics, concentration and fuel-cell measurements, and impedance spectroscopy [J]. Solid State Ionics, 1999, 107: 107-117.
[84] SCHOBER T, BOHN HG, MONO T, et al. The high temperature proton conductor Ba3Ca1.18Nb1.82O9-δ; II, electrochemical cell measurements and TEM [J]. Solid State Ionics, 1999, 118: 173-178.
[85] SCHOBER T, KRUG F, SCHILLING W. Criteria for the application of high temperature proton conductors in SOFCs [J]. Solid State Ionics, 1997, 97: 369-373.
[86] BOHN HG, SCHOBER T, MONO T, et al. The high temperature proton conductor Ba3Ca1.18Nb1.82O9-δ: I, electrical conductivity [J]. Solid State Ionics, 1999, 117: 219-228.
[87] MURRAY EP, TSAI T, BARNETT SA. Oxygen transfer processes in (La,Sr)MnO3/Y2O3-stabilized ZrO2cathodes: an impedance spectroscopy study [J]. Solid State Ionics, 1998, 110: 235-243.
[88] VALKENBERG S, BOHN HG, SCHILLING W. The electrical conductivity of the high temperature proton conductor Ba3Ca1.18Nb1.82O9-δ[J]. Solid State Ionics, 1997, 97: 511-515.
[89] 田力, 董笑, 田野. 掺杂氧化铈-碳酸盐燃料电池复合电解质[J]. 电源技术, 2014, 38 (7): 1374-1398.
[90] ZHU B, Liu X, ZHOU P, et al. Innovative solid carbonate-ceria composite electrolyte fuel cells [J]. Electrochemistry Communications, 2001, 3 (10): 566-571.
[91] JING Y, MA Y, PATAKANGAS J, et al. Enhanced conductivity of SDC based nanocomposite electrolyte by spark plasma sintering [J]. International Journal of Hydrogen Energy, 2014, 39 (26): 14391-14396.
[92] XIA C, LI Y, TIAN Y, et al. A high performance composite ionic conducting electrolyte for intermediate temperature fuel cell and evidence for ternary ionic conduction [J]. Journal of Power Sources, 2009, 188 (1): 156-162.
[93] HUANG J, MAO Z, LIU Z, et al. Development of novel low-temperature SOFCs with co-ionic conducting SDC-carbonate composite electrolytes [J]. Electrochemistry Communications, 2007, 9 (10): 2601-2605.
[94] ZHAO Y, XU Z, XIA C, et al. Oxide ion and proton conduction in doped ceria–carbonate composite materials [J]. International Journal of Hydrogen Energy, 2013, 38 (3): 1553-1559.
[95] WU G, WANG C, XIE F, et al. Ionic transport mechanism of La0.9Sr0.1Ga0.8Mg0.2O2.85-(Li/Na)2CO3composite electrolyte for low temperature SOFCs [J]. International Journal of Hydrogen Energy, 2016, 41 (36): 16275-16281.
[96] VENKATASUBRAMANIAN A, GOPALAN P, PRASANNA TRS. Synthesis and characterization of electrolytes based on BaO–CeO2–GdO1.5system for intermediate temperature solid oxide fuel cells [J]. International Journal of Hydrogen Energy, 2010, 35 (10): 4597-4605.
[97] KHANDELWAL M, VENKATASUBRAMANIAN A, PRASANNA TRS, et al. Correlation between microstructure and electrical conductivity in composite electrolytes containing Gd-doped ceria and Gd-doped barium cerate [J]. Journal of the European Ceramic Society, 2011, 31 (4): 559-568.
[98] SUN W, JIANG Y, WANG Y, et al. A novel electronic current-blocked stable mixed ionic conductor for solid oxide fuel cells [J]. Journal of Power Sources, 2011, 196 (1): 62-68.
Research Progress on Electrolytes for Intermediate-Temperature Solid Oxide Fuel Cells
SUN Hai-Bin1, GUO Xue1, ZHANG Zhen-Hao2, ZHANG Yu-Jun3
1School of Materials Science and Engineering, Shandong University of Technology, Zibo 255049, China2Laiwu Advanced Ceramic Technology Co., Ltd, Laiwu 271100, China3School of Materials Science and Engineering, Shandong University, Jinan 256216, China
The electrolyte is one of the core components for solid oxide fuel cell (SOFC), which is a new energy conversion device. To promote the development of intermediate-temperature SOFC, the electrolytes should have high ionic conductivities under medium temperature conditions (500oC ~ 700oC) and have good long-term stabilities in the fuel cell operating environment. In this paper, the research progresses of oxygen ion conductive electrolytes, proton conductive electrolytes and composite electrolytes are reviewed, and the difficulties inhibiting their development and applications are analyzed. Finally, the development trends of medium temperature electrolytes are proposed.
Solid oxide fuel cell; Oxide-conduction electrolyte; Proton-conduction electrolyte; Composite electrolyte.
TB333
1005-1198 (2018) 06-0403-14
A
10.16253/j.cnki.37-1226/tq.2018.07.010
2018-07-14
2018-10-21
国家自然科学基金 (51702189); 山东省自然科学基金 (ZR2017BEM033); 山东省高校科研计划 (J18KA002)。
孙海滨 (1984-), 男, 山东滨州人, 讲师。E-mail: hbsun@sdut.edu.cn。
孙海滨,男,1984年1月生,讲师,山东理工大学“双百工程第三层次人才”,主要从事固体氧化物燃料电池、结构陶瓷研究。近年来,致力于将微波烧结、热压烧结、大气等离子喷涂等陶瓷制备技术应用于固体氧化物燃料电池,实现其低成本制备,并研究晶界特性对陶瓷电解质电导性能的影响。截至目前,主持国家自然科学基金1项、山东省自然科学基金1项,已发表学术论文30余篇,获得授权国家发明专利5件,获省部级科技进步二等奖3项、市级科技进步一等奖3项。

