分子动力学模拟在推进剂组分物理化学性能研究中的应用进展
2019-01-19张崇民赵小锋付小龙樊学忠李吉祯
张崇民,赵小锋,付小龙,樊学忠,李吉祯
(1.西安近代化学研究所,陕西 西安 710065;2.海军装备部驻西安地区军事代表局,陕西 西安 710054)
引 言
固体推进剂的主要组分包括氧化剂、黏合剂、增塑剂、金属颗粒、固化剂和键合剂等。其中氧化剂、黏合剂与金属颗粒可为推进剂提供能量;增塑剂、固化剂和键合剂可辅助推进剂固化成型,并改善其力学性能[1]。在实际使用中,几十年来,对推进剂配方的选取及其性能的研究,需经过大量实验进行筛选,这将造成人力、物力、财力的巨大浪费。而利用分子动力学方法(MD)对推进剂组分进行模拟,可以从微观的角度研究其结构和性能之间的关系,并进一步指导推进剂的配方设计[2],提高研究效率。
随着现代蒙特卡罗方法的成功应用[3],国外研究者[4]在此基础上,模拟了硬球模型的弹性碰撞,发展了分子动力学方法。该方法把原子近似为质点或点电荷,以库仑力或者范德华力等受距离影响的函数描述其相互作用。化学键则是由弹簧力来模拟,官能团被参数化以模拟由量子力学提供的势能面,该参数集一般由Materials Studio软件中的COMPASS力场提供。在该软件中,可在Forcite模块中优化构建的分子模型,并分配其力场类型。同时,可以创建一个体积空间,模拟大量的分子原子,如晶体、液体或密集的分子进行填充。最后采用经典运动方程与原子间力的迭代求解可模拟出体积元素内结构和能量的演化。
本文从推进剂的主要组分出发,综述了分子动力学模拟方法在固体推进剂研究中的应用,总结了分子动力学方法对推进剂组分的微观结构、热分解及力学性能等特性的模拟。
1 分子动力学方法在固体推进剂中各组分的应用进展
1.1 氧化剂
在推进剂中,氧化剂所占含量的比例较大,双基推进剂常用的氧化剂有RDX、HMX和CL-20,以及复合推进剂常用的氧化剂AP、ADN和AN。
HMX有4种晶型,分别为α-HMX、β-HMX、γ-HMX和δ-HMX。通过对这4种晶型的模量和升华热等性质的模拟发现,β-HMX的模量最大[5],且升华焓最高[6]。对其晶胞参数的模拟如表1所示,可以看出理论计算与实验结果吻合良好。

表1 β-HMX的晶胞参数和密度的MD计算值与实验值的比较[7]Table 1 Comparison of the calculated values and the experimental ones by MD for crystal cell parameters and density of β-HMX
在建立HMX晶体模型时,以β-HMX为例[8],需考量超晶胞大小和形状以及是否切割分面和切割深度等模型建立问题。在考虑晶体缺陷情况下,构建HMX纯晶体、含质量分数5.6%RDX和含质量分数为5.6%空穴的HMX晶体的3种模型[9],并对其力学性能做了模拟,结果如表2所示。
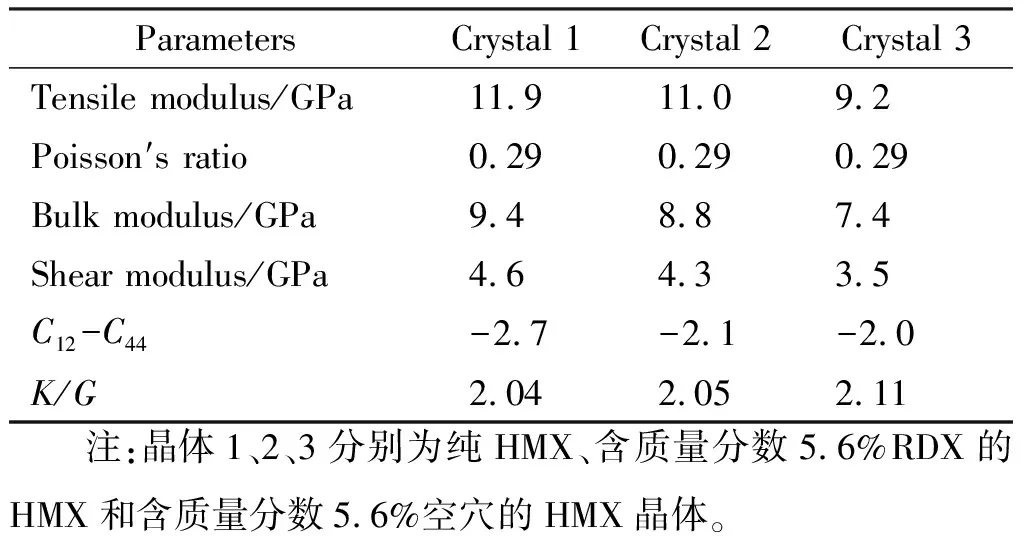
表2 HMX纯晶体和HMX缺陷晶体的力学性能Table 2 Mechanical properties of perfect HMX and defect HMX crystal
由表2可知,含质量分数5.6%RDX和含5.6%空穴的HMX晶体的弹性系数和模量均小于HMX纯晶体,而其柯西压(弹性系数C12与C44的差值)和K/G(体积模量和剪切模量之比)增大,这表明HMX晶体的脆性减弱,韧性和延展性增强。因此一定程度的晶体缺陷会影响其力学性能,这对推进剂的配方设计有一定的指导意义。
HMX在高温受热之后会发生热分解,其δ构型如图1所示。
由图1可见,在晶胞中有6个HMX分子,应用分子反应动力学通过ReaxFF力场对该模型研究表明,在2500K高温下,其主要分解机理为N—NO2键断裂和HONO的消去;在1500K温度下,其主要分解机理为N—NO2键断裂和环的断裂[10-11]。通过对N—N键的分解模拟研究表明[12],直链硝胺比环状硝胺分解快,因此在实际应用中,可使用低感度环状硝胺,如β-HMX、RDX等。当HMX受到冲击压缩时,采用三阶Birch-Murnaghan方程和Hugoniot关系拟合[13-14],随着体系温度升高,β-HMX相比于其他晶型更容易压缩,且晶胞的压缩具有各向异性,这种压缩异性不随温度的改变而改变,并且可以通过对单轴压缩的模拟来研究单晶炸药的冲击响应,计算HMX晶体的绝热压缩来预估炸药的冲击温度[15]。
偏二氟乙烯与三氟氯乙烯摩尔比1∶1的共聚产物(F2311)对HMX的性能有一定的改善作用[16-19],在COMPASS力场下,模拟结果如表3所示,氟聚物的加入会提高体系的弹性、延展性,改善体系力学性能,并且HMX(1 0 0)晶面与F2311之间的柯西压最大,即韧性最好。对HMX/F2311体系的爆热和爆速的模拟如表4所示,在加入F2311之后,HMX体系的爆热和爆速下降,安全性有所提高。

表3 纯HMX和HMX/F2311的弹性系数和有效各向同性力学性能[17] Table 3 Elastic coefficient and effective isotropic mechanical properties for pure HMX and HMX/ F2311[17]

表4 HMX和HMX/F2311的爆热和爆速Table 4 The heat of detonation and detonation velocity of HMX and HMX/F2311
在HMX力学性能的分子动力学模拟研究方面,研究者主要应用COMPASS力场,对部分HMX体系的力学性能做了计算。TATB[18, 20]、NC[19]、PEG、HTPB[21]和Estane[22]等均表明可以改善HMX体系的力学性能,提高HMX体系的延展性,且NC还可以增强体系韧性,提高能量性能。
结合能的计算公式如下:
Ebinding=-Einter=-(EA/B-EA-EB)
(1)
式中:EA/B为A和B混合体系的总能量;EA和EB则分别为A和B的总能量。
ZHU Wei等[23]对AP与HMX之间的结合能进行研究,通过模拟得到ETotal、EAP和EHMX,并计算出结合能先随温度的上升而增加,之后下降,温度达到245K时,结合能最大,可达962.9kJ/mol。
对RDX力学性能的模拟表明[24],在195~345K下,RDX延展性和韧性随温度升高而增大;在345~445K下,RDX延展性和韧性随温度升高而减小,呈抛物线规律。对RDX晶体的晶胞参数、质量分数中心、欧拉角度和晶格尺寸等晶体基本参数的模拟[25],有助于对RDX进行深入研究。模拟RDX晶体缺陷对其最大引发键键长的影响的结果如表5所示[26],有一个空位缺陷的RDX晶体的最大引发键键长最大,即感度最高。因此,晶体缺陷会对推进剂的稳定性产生一定的影响。

表5 (1 0 0)RDX的完美和缺陷晶体的N-NO2引发键的Lmax和LaveTable 5 Lmax and Lave for the N-NO2 trigger bonds in perfect and defective (1 0 0) RDX
RDX也可作为Al的包覆材料[27],RDX中的氧原子和Al原子之间的强作用力会导致RDX中的N-N和N-O键分解,且Al的(1 1 1)晶面会被氧化,而剩下的RDX片段则会吸附在Al原子的表面上阻止Al的进一步氧化。向RDX中添加聚硫橡胶(PS)之后[28],随温度升高,RDX引发键最大键长Lmax增大,引发键(N—NO2)中EN-N减小,内聚能密度减小。研究GAP接枝二甲基海因(GAP-DMH)与RDX晶体之间的相互作用的结果表明[29],GAP-DMH在RDX晶体不同晶面上的结合能大小为(0 0 1)>(0 1 0)>(1 0 0),且因为GAP-DMH与RDX晶面之间的距离更接近,所以相比于 GAP/RDX体系拥有更大的范德华力,即有更高的结合能。
许多学者研究了AP颗粒为填料来构建固体推进剂颗粒填充模型,如图2所示,分子在运动中会不断碰撞,导致直径增大,最终颗粒的填充体积分数会达到预期要求,以计算推进剂的某些性能参数,如弹性模量、泊松比和导热系数等[30-32]。1990年,Lubachevsky[33-34]首次提出使用分子动力学方法来生成颗粒填充模型的算法,之后Knot[35]将这种办法成功用于复合固体推进剂体系中。
李高春等[36]基于此模型对复合固体推进剂中的颗粒分布进行模拟,并应用细观力学Mori-Tanaka方法对推进剂的杨氏模量做计算,结果如图3所示,实验值与计算值吻合较好。
张建伟等[37]构建以AP颗粒为填料的固体推进剂颗粒填充模型,模拟该体系模型的松弛模量曲线,结果表明,AP颗粒主要增强了体系的瞬时模量,且AP颗粒体积分数越大,增强效果越强,瞬时模量增加速度越高。采用此模型还可对推进剂的微观损伤做出模拟预测,职世君等[38-41]使用Surface-based Cohesive方法模拟了固体颗粒与基体填料体系中的损伤,得到了界面损伤形貌,使用迭代法计算出了合理的界面损伤参数,并模拟分析了界面损伤初始应力、初始刚度和界面失效距离对复合固体推进剂最大延伸率的影响,结果表明,受固体推进剂细观颗粒的影响,AP颗粒含量增加,推进剂的最大延伸率会减小,形成的宏观裂纹会越明显并有一定的随机性,且界面损伤多出现在大颗粒附近。通过对感度的计算,还可筛选推进剂的配方,研究AP/HMX体系的不同配比与感度的关系,结果如表6所示,当AP/HMX摩尔比为1∶1时,其感度最大[42],N—NO2平均键长(LN-NO2)可达1.373×10-1nm,最大引发键键长(Lmax)可达1.510×10-1nm,而在该体系中加入黏合剂后,如(PEG/NG/BTTN)/AP/HMX体系,当其摩尔比为2.5∶3.5∶2.3时,感度相对最大[43]。
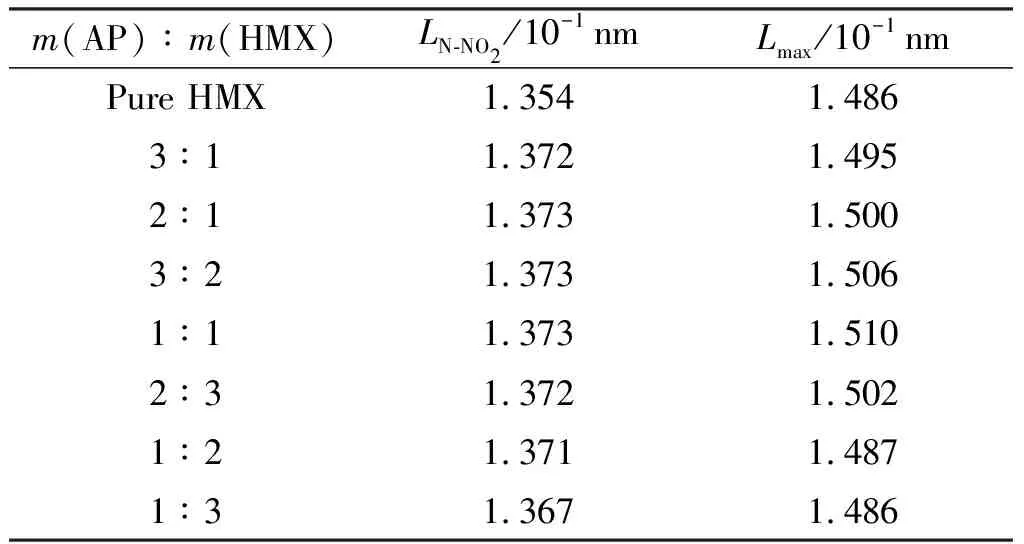
表6 不同质量比AP/HMX中N-NO2的平均键长(LN-NO2)和最大键长(Lmax)Table 6 Average(LN-NO2)and maximal(Lmax)bond lengths of N-NO2 in AP/HMX with different mass ratios
CL-20为笼型多环硝胺结构,是一种白色结晶,常使用稳定性最好的ε-CL-20晶型[44]。在COMPASS力场下模拟温度升高,发现ε-CL-20中引发键(N-NO2)最大键长Lmax递增,EN-N、内聚能密度(CED)递减,即感度随温度升高而增大[45]。当ε-CL-20与黏合剂混合时,通过结合能和径向分布函数的模拟,可研究黏合剂的加入对ε-CL-20的影响,结果表明,氟聚物与ε-CL-20相容性较差,而PEG、HTPB和GAP均与ε-CL-20相容性较好,且黏合剂的加入能提高ε-CL-20体系的延展性,并保持体系密度大于1.9g/cm3[46]。当其与HMX混合时,模拟结果表明,体系的感度和延展性都优于其单体[47]。
以上研究表明,分子动力学方法可以对氧化剂的力学性能、分子间相互作用等性质做出较好的模拟,并对其感度做出初步预测。同时,从氧化剂的分子动力学研究趋势可以看出,国内外研究者逐渐由对氧化剂微观结构的模拟向氧化剂具体性能的模拟转变,由单一组分模拟向复杂组分的模拟转变。
1.2 黏合剂
HTPB抗老化性能好,储存寿命长且黏度低,是目前在固体推进剂中得到广泛应用的黏合剂[48],其无定型结构如图4所示。
马昌兵等[49]根据图4中的结构,将HTPB与AP和Al混合之后,得到的新结构具有各向同性、统计均匀性和各态历经性。李金龙等[50]在COMPASS力场下,可模拟不同溶剂与HTPB基聚氨酯之间的结合能和扩散系数,比较HTPB基聚氨酯的溶胀率,其结果如表7所示,溶剂对黏合剂会有一定的溶胀作用,三氯甲烷对HTPB基聚氨酯的溶胀率最高,与图5完全吻合。李红霞等[51]采用COMPASS力场,还可通过模拟HTPB分子的比体积-温度曲线,计算 HTPB的玻璃化温度,但是模拟结果相比于实验值偏大,可能是因为模拟中的冷却速度更快[52]。
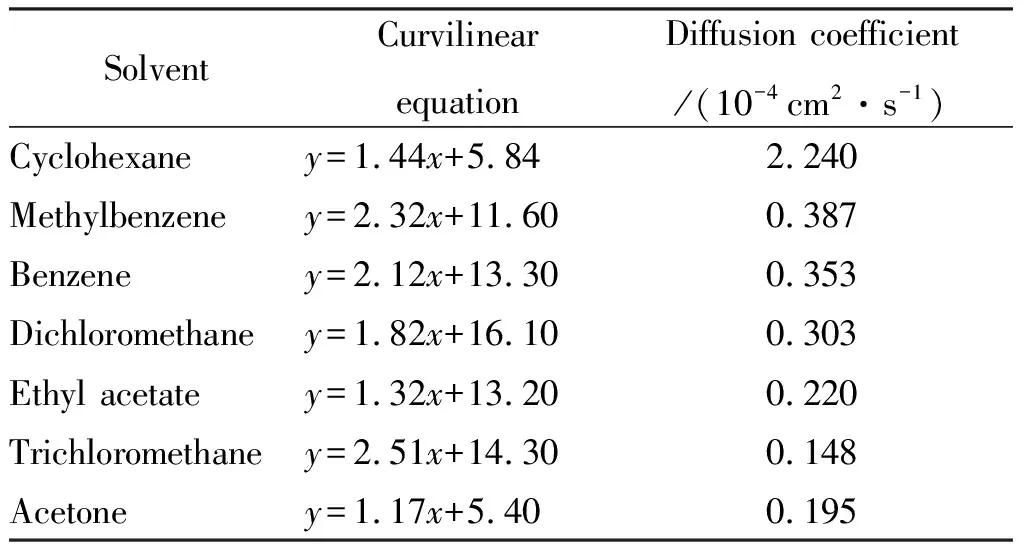
表7 HTPB在不同溶剂中的扩散系数模拟值Table 7 The simulated values of diffusion coefficient for HTPB in different solvents
齐晓飞等[53]采用分子动力学方法,根据NC的模型,将NG分子与NC分子混合在一起,计算NG对NC分子回转半径的影响,结果如表8所示,升高温度会有限地提高回转半径,且NG越多,回转半径越大,最大回转半径为2.618nm,通过径向分布函数的模拟,发现这是因为NG中—ONO2基团与NC中—OH之间的氢键弱化了NC分子内的氢键,所以NC分子链的回转半径增大。黄锐等[54]比较了硝化纤维素(NC)、硝化纤维素甘油醚(NCEC)之间力学性能及加工性能,发现 NCEC双基推进剂要优于NC双基推进剂。

表8 各模型中NC分子链在不同温度下的回转半径Table 8 Radius of gyration for NC at different models and temperatures
以上研究表明,分子动力学方法可以对黏合剂的分子链构象及其与溶剂之间的溶胀性质做出较好的预测,这对黏合剂在固体推进剂中应用的选择提供了理论指导,但多组分体系仍需进一步研究。
1.3 增塑剂
推进剂组分与增塑剂之间的相溶性对推进剂的力学性能影响较大。当判断组分间的相溶性时,可使用溶解度参数、内聚能密度等性能参数,采用Materials Studio中的Synthia方法和Blend方法可以得到相关结果[55]。黏合剂与增塑剂的溶解度参数的模拟如表9所示,当|Δδ|<1.3~2.1(J·mol-3)1/2时认为两者相溶性较好,根据表9结果可知,HTPB/DOS、HTPB/DBP、HTPB/DEP体系相溶,而HTPB/DEGDN[56]、HTPB/NG[57]体系不相溶,其中HTPB/DOS无定形分子模型如图6所示,将该模型放在COMPASS力场下计算,当DOS质量分数为2%~4%,温度高于228K时[58],其扩散系数最大,共混效果最好[59-61]。对端羟基聚醚(HTPE)与N-丁基硝氧乙基硝胺(Bu-NENA)之间的相溶性的模拟结果表明[62],Bu-NENA的溶解度参数为19.687(J·mol-3)1/2,HTPE的溶解度参数为19.034(J·mol-3)1/2,即|Δδ|=0.653(J·mol-3)1/2,故该体系相溶。

表9 黏合剂与增塑剂的溶解度参数Table 9 Solubility parameters of the adhesive and plasticizer
虞振飞等[63]采用分子动力学方法模拟推进剂体系中组分迁移,研究NG与BTTN在聚氨酯弹性体中的迁移,结果表明,随温度升高,NG和BTTN在聚氨酯中扩散系数增大,且NG的扩散系数大于BTTN。模拟DOS和NG的加入对HTPB体系的玻璃化温度的影响结果如图7所示[52,64],在加入DOS和NG之后,体系原有的玻璃化温度会降低,这与实验观测到的结果一致。李晓颖等[65]进一步用力场能量项分析得到玻璃化温度与二面角扭转能正相关。
以上研究表明,分子动力学方法可以对增塑剂与黏合剂之间混溶性做出良好的预测,有助于对增塑剂的选取和对推进剂配方的研究,但计算精确度仍需进一步提高。
1.4 金属颗粒
金属颗粒在固体推进剂中一般作为高能燃烧剂使用,在燃烧时会释放出大量的热能,提高了推进剂的燃烧温度,从而提高理论比冲与特征速度。常用的金属颗粒为铝粉、镁粉等,从微观上看,铝颗粒在推进剂中会作为分散相存在,而黏合剂则会作为连续相存在,所以铝颗粒与黏合剂之间的粘结状况会对固体推进剂的力学性能有显著影响。兰艳花等[66]以聚乙二醇(PEG)为黏合剂时,在COMPASS力场下,建立了高能推进剂NEPE的组分PEG/Al球形包覆模型,力学性能模拟结果如表10所示,可见Al的加入对PEG的拉伸、剪切模量影响不大。

表10 PEG/Al球形包覆体系的力学性能Table 10 Mechanical properties of PEG/Al globular coated system
由泊松比μ<0.2可知,加入Al之后PEG体系韧性较好,具备某些塑料特性。计算其结合能EB为1463.66kJ/mol,而PEG的能量EP为-22118kJ/mol,故PEG的分子内能量比PEG/Al之间的结合能小的多,所以,在PEG分子中容易出现分子内链断裂,而Al和PEG之间不易脱粘。
付一政等[67]以HTPB为黏合剂时,其力学性能结果如表11所示,HTPB与Al(0 1 1)晶面之间的结合能最大,同时HTPB沿Al(0 1 1)晶面综合力学性能最好。
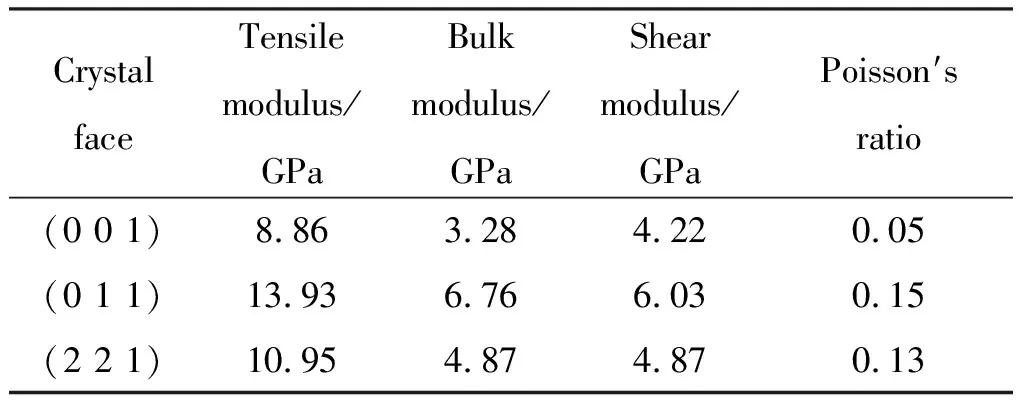
表11 400K下HTPB/Al的力学性能Table 11 Mechanical properties of HTPB / Al at 400K
以上研究结果表明,通过分子动力学模拟,可以计算金属颗粒与黏合剂之间的结合能与力学性能,从而判断黏合剂/金属颗粒体系的稳定性,但多组分体系仍需进一步研究。
1.5 固化剂
黏合剂与固化剂之间的固化体系对推进剂整体的力学性能有着十分重要的关系。常用的固化剂是甲苯二异氰酸酯(TDI)、异佛尔酮二异氰酸酯(IPDI)和六亚甲基二异氰酸酯(HDI)等。采用分子动力学方法,可对推进剂固化反应的机理做出预测。HTPB/TDI体系的研究结果表明[68],该体系在固化过程中,TDI的氰酸酯基(—NCO)中的N=C变成单键,羟基O—H断裂生成C—O键和N—H键,对其C—O键变化的模拟结果表明,固化过程中C—O键键长会不断减小,直到固化完成。
当推进剂开始老化时,利用Materials Studio中的Dmol3模块研究发现,HTPB/TDI结构中与—CH2相连的的C—O键最容易断裂,其结构如图8所示,断裂仅需244.95kJ/mol的能量[69]。在体系老化之后,体系的弹性模量会增加,因为在老化中释放的CO2会扩散聚集[70],从而使体系形成局部有空隙的聚合体结构,该结构相比于未老化结构,拉伸模量和剪切模量会上升。
在传统内聚力中引入黏弹性因素,对传统的计算模型做出修正,构建考虑率效应的HTPB/IPDI内聚力模型[71],并做参数反演优化,结果表明,颗粒随机分布对材料力学性能无影响,且可有效体现出HTPB/IPDI推进剂力学性能的应变率相关性。
以上研究表明,分子动力学方法可以对黏合剂与固化剂之间的固化机理做出较好的模拟,可预测固化体系的老化过程中的结构及性能变化,但对其动力学过程需进一步深入研究。
1.6 键合剂
固体颗粒和黏合剂之间的粘结状况是影响推进剂力学性能的关键因素,为了防止推进剂出现脱湿的状况,可在固体推进剂中添加键合剂[72],采用分子动力学方法,可计算键合剂在金属颗粒和黏合剂上的吸附情况,如图9所示,在298K下,三(-2甲基氮丙啶-1)氧化磷(MAPO)的尾端会吸附在Al2O3上,且在Al2O3(0 1 0)晶面上的吸附能最大可达13199kJ/mol。模拟不同键合剂在HTPB(端羟基聚丁二烯)和Al/Al2O3上的吸附的结果如表12所示[73-75],键合剂在Al2O3上的(0 1 0)面吸附能最大,对力学性能的模拟表明,随键合剂摩尔浓度增大,体系的弹性模量也增大,并指出TAZ(k-氮丙啶丙酸与2, 2-二羟基丁醇反应生成的三酯的简称)对体系力学性能改善最好[76],而且加入键合剂之后,还可以减少氧化剂分解气体O2、H2O在键合剂膜层的扩散能力和改善推进剂贮存抗老化性能。

表12 键合剂在晶面和HTPB界面的吸附能Table 12 Adsorption energy of different bonding agents on crystal faces and HTPB surfaces
采用分子动力学,还可模拟接枝海因对黏合剂的影响,模拟聚叠氮缩水甘油醚(GAP)和GAP与3-炔丙基-5,5-二甲基海因(PDMH)的反应产物GAP-PDMH与RDX、HMX和AP构成的体系,结果表明[77],因为在PDMH上的三唑基团与海因基团与氧化剂之间有更强的相互作用力,所以GAP-PDMH与氧化剂之间有更大的相互作用能。
以上研究表明,分子动力学方法可以对键合剂与黏合剂之间的结合能做出预测,有助于键合剂的选择和推进剂配方的调整,从而解决推进剂的脱湿问题,但多组分体系仍需进一步研究。
2 结束语
分子动力学方法可模拟固体推进剂组分的微观结构,从而预测其宏观性能。因此,分子动力学方法的发展将对固体推进剂的深入研究有着重要影响,也将成为固体推进剂理论设计的主要方法之一。目前,分子动力学方法在固体推进剂中的应用仍需解决以下几方面的问题:
(1)在研究对象的选择方面,目前主要开展了固体推进剂中单一组分的研究,对多组分体系乃至全组分的研究尚不深入或空白,因此,亟需开展固体推进剂多组分体系的分子动力学研究。
(2)在结构与性能研究方面,对固体推进剂的微观结构与其宏观性能之间内在关系的研究尚不透彻,且计算精度有待提高,因此,亟需开展推进剂结构与性能间的深入研究,并加强实验验证。
(3)在动力学研究方面,对固体推进剂的固化反应、分解反应和燃烧反应等反应历程的研究基本尚未开展,因此,对固体推进剂的分子反应动力学研究将是固体推进剂的重点研究方向。
