Exploratory clinical study of chidamide, an oral subtype-selective histone deacetylase inhibitor, in combination with exemestane in hormone receptor-positive advanced breast cancer
2019-01-18QingyuanZhangTaoWangCuizhiGengYueZhangJinwenZhangZhiqiangNingZefeiJiang
Qingyuan Zhang, Tao Wang, Cuizhi Geng, Yue Zhang, Jinwen Zhang, Zhiqiang Ning, Zefei Jiang
1Department of Medical Oncology, Harbin Medical University Cancer Hospital, Harbin 150081, China; 2Department of Breast Cancer, the 307th hospital of Chinese People’s Liberation Army, Beijing 100071, China; 3The Fourth Hospital of Hebei Medical University, Shijiazhuang 050011,China; 4Shenzhen Chipscreen Biosciences, Shenzhen 518057, China
Abstract Objective: The recurrence or progression under endocrine therapy in hormone receptor-positive (HR+)advanced breast cancer (ABC) remained a critical clinical challenge. Chidamide is an oral subtype-selective histone deacetylase (HDAC) inhibitor with multiple functions in tumor growth inhibition and microenvironment modulation via epigenetic reprogramming. The purpose of this study was to evaluate the safety, pharmacokinetics(PK), and preliminary efficacy of chidamide in combination with exemestane in HR+ ABC patients.Methods: Eligible patients were postmenopausal women with HR+ ABC recurrent or progressed to at least one endocrine therapy. Blood samples were obtained in the run-in period and the first day of combination treatment for PK analysis. In combination treatment, patients were given exemestane 25 mg daily and chidamide 30 mg twice a week (BIW) until progression of disease or intolerable toxicities. A treatment cycle was defined as 4 weeks. Safety,PK parameters, and preliminary efficacy were evaluated.Results: A total of 20 patients were enrolled between July and December, 2015. The median number of treatments cycle was 5.2 (20.8 weeks) with 2 patients still on treatment at the data cut-off date of October, 2017.The treatment-related adverse events (AE) ≥ grade 3 in more than 2 patients were neutropenia (35%),thrombocytopenia (30%), and leucopenia (20%). The plasma exposure of exemestane was consistent in the presence or absence of chidamide. A slight increase in chidamide exposure was noted in the presence of exemestane,probably due to the inter- and intra-patient variations. The best response in 16 evaluable patients was assessed by Response Evaluation Criteria in Solid Tumors (RECIST), including 4 patients with partial response, 10 patients with stable disease. The median progression-free survival (PFS) was 7.6 months.Conclusions: The combination of chidamide with exemestane was generally well tolerated with promising preliminary efficacy in HR+ ABC patients. The overall results from this study encourage further pivotal trial in this patient population.
Keywords: Advanced breast cancer; hormone receptor-positive; chidamide; exemestane
Introduction
It is shown that 50%-70% of breast cancers are estrogen receptor (ER) positive and/or progesterone receptor (PR)positive (1). Endocrine therapies are the foundation for treatment of these hormone receptor-positive (HR+)cancers. Despite advances in endocrine therapy, either primary or secondary drug resistance is a common issue in patients with the treatment, leading to disease progression or recurrence (2). Although the intrinsic mechanisms of endocrine resistance are incompletely uncovered, ER dysfunction (silence, hypersensitivity, mutation, etc.),alternative activation of growth signaling pathways, and/or altered epigenetic modifications are suggested by preclinical and clinical studies (3-5).
Histone deacetylase (HDAC) inhibitors are a promising drug class in overcoming resistance to endocrine therapy.In general, those compounds induce cell proliferation arrest, differentiation, and cell death in breast cancer cells(6). Also, HDAC inhibitors, such as vorinostat and entinostat, have been shown to restore hormone sensitivity via down-regulation of estrogen-independent growth factor signaling pathways, normalization of ER levels, and increase in aromatase enzyme levels in vitro and preclinical studies (7-9). Furthermore, in a randomized phase II trial the addition of entinostat to exemestane showed a potential prolonged progression-free survival (PFS) and overall survival (OS) in patients with previously treated hormonesensitive metastatic breast cancer (10).
Chidamide (CS055/Tucidinostat/Epidaza®) is an oral benzamide class of HDAC inhibitor with broad antitumor activities by selectively inhibiting HDAC1, 2, 3 and 10 (11),and has been approved by the China Food and Drug Administration (CFDA) as a treatment for relapsed or refractory peripheral T cell lymphoma (PTCL) (12).Previous studies have shown that benzamide class of HDAC subtype-selective inhibitors, including chidamide and entinostat, enhance tumor immune surveillance via activation of natural killer (NK) cell and antigen-specific cytotoxic T lymphocyte (CTL)-mediated cellular antitumor immunity, which differentiates them from other non-selective HDAC inhibitors (13-16). Chidamide has also been demonstrated to down-regulate estrogenindependent growth factor signaling pathways and restore the sensitivity to anti-estrogen agents in preclinical investigations (17).
Exemestane is a steroidal aromatase inhibitor (AI) with well-established efficacy in HR+, human epidermal growth factor receptor 2 (HER2)-negative advanced breast cancer(ABC) patients progressed on previous endocrine therapy,including tamoxifen and/or nonsteroidal AIs (i.e., letrozole or anastrozole). This exploratory clinical study was thus performed to evaluate the safety, pharmacokinetics (PK),and preliminary efficacy of chidamide in combination with exemestane in postmenopausal women with HR+ and HER2-negative ABC that recurrent or progressed to at least one endocrine therapy.
Materials and methods
Study design
The study design was a single-arm, open-label, exploratory trial of chidamide in combination with exemestane in HR+ABC patients. The Institutional Review Board at each participating center approved the study, which was conducted by the principles of Good Clinical Practice, the provisions of the Declaration of Helsinki, and other applicable local regulations. The study was registered on ClinicalTrials.gov (No. NCT02482753).
The primary objective was to determine the safety and PK profiles of chidamide with exemestane in ABC patients.The secondary goal was to evaluate the preliminary efficacy, including objective response rate (ORR) and PFS.
In the 4-day-run-in period, patients received singleagent exemestane 25 mg (provided by Pfizer Inc.) on d 1 and chidamide 30 mg (supplied by Shenzhen Chipscreen Biosciences, Ltd.) on d 2. From d 5, patients started combination treatment with exemestane 25 mg daily and chidamide 30 mg twice a week (BIW). A treatment cycle was defined as 28 d.
Before study entry, patients underwent a complete history and physical examination, and all prior anticancer treatments and their residual side effects were recorded, if any. Baseline imaging evaluations were obtained to define the extent of disease. Baseline laboratory tests included a complete blood cell, platelet counts and blood chemistry.Physical examination, laboratory assessments and radiological evaluations were repeated every 8 weeks.Toxicities were graded using the National Cancer Institute Common Toxicity Criteria (NCICTC), version 4.0.
Patients continued on treatment until they had disease progression, or were unable to tolerate with therapy, or withdrew consent.
Patients and eligibility criteria
Written informed consent was obtained from all patients before enrollment. Eligible patients included: 1) 18 to 75 years old postmenopausal women and had an Eastern Cooperative Oncology Group performance status of 0 or 1;2) ER+, HER2-negative advanced or metastasis breast cancer patients who were experiencing disease relapse or progression after at least one endocrine therapy; 3) only one prior line of chemotherapy in the metastatic setting was permitted; 4) the total number of endocrine therapy and chemotherapy did not exceed 4 regimens; and 5)disease had to be measurable by Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 or nonmeasurable bone-only disease.
Exclusion criteria mainly included 1) prior treatment with exemestane; 2) presence of visceral crisis; 3) evidence or history of central nervous system (CNS) metastasis; 4)cancer chemotherapy, radiation therapy, or any investigational agent within 4 weeks before the baseline evaluations; 5) an active severe infection; 6) unstable angina within 6 months; 7) myocardial infarction within 12 months; 8) therapy for life-threatening ventricular arrhythmia; or 9) life-threatening visceral disease or other severe concurrent diseases.
Estrogen, progesterone, and HER2/neu receptor status in tumors were obtained from patient histories when available. HER2/neu receptor status could be determined by immunohistochemical analysis or fluorescence in situ hybridization.
Safety was assessed via adverse events (AEs), vital signs,electrocardiogram, and laboratory tests. The investigator determined AE seriousness, severity grade, and relationship to study treatment. AEs were graded by the NCICTC for Adverse Events version 4.0.
Tumor response was assessed using RECIST Version 1.1. Magnetic resonance imaging or computed tomography scans were performed at screening, 8 weeks after the first dose of study treatment, and every 8 weeks after that until documented radiographic progression, the initiation of other anticancer therapy or death. The number of patients with complete response (CR), partial response (PR), stable disease (SD), and progressive disease (PD) were determined.
Pharmacokinetic sampling and analysis
For exemestane, plasma samples were collected at pre-dose and 1, 2, 4, 8, 12 and 24 h post-dose on d 1 of the run-in period and d 1 of combination treatment, respectively. For chidamide, plasma samples were collected at pre-dose and 1, 2, 4, 8, 12, 24, 48 h and 72 h post-dose on d 2 of the runin period and d 1 of combination treatment, respectively.All plasma samples were stored at -10 °C to -30 °C until analysis.
A validated high-performance liquid chromatographytandem mass spectrometry (HPLC-MS/MS) method was used to determine the plasma concentrations of chidamide(18,19) by Covance Analytical Laboratory (Shanghai,China). Plasma concentrations of exemestane were determined by Covance Laboratory from its internal validated method. The calibration curve range for chidamide and exemestane was 1-500 ng/mL and 0.2-100 ng/mL, respectively.
Statistical analysis
Descriptive statistics, such as mean, median, frequency and proportion, were used to summarize the results along with 95% confidence intervals (95% CI) where applicable. The Kaplan-Meier method was used to estimate PFS statistics.No inferential statistical analyses were performed due to its nature as a single-arm trial. All statistical analyses were performed using SAS software (Version 9.2; SAS Institute Inc., Cary, NC, USA).
The maximum plasma drug concentration (Cmax) and time to reach maximum concentration (Tmax) were obtained from experimental observation. Other PK parameters, including t1/2, area under the curve (AUC)0-t,AUC0-∞, clearance (CL/F), volume of distribution (Vd/F),and mean residence time (MRT), were calculated by a noncompartmental model with WinNonlin v6.4 (Pharsight Corp., Mountain View, CA, USA).
Results
Patient characteristics and disposition
Between July 2015 and December 2015, 20 eligible patients were recruited, and their baseline characteristics are listed in Table 1. Most patients were in late middle age, and the median age was 56 years old. In this study, 35% of the patients had visceral involvement, and 30% had bone-only metastasis; 70% of the patients had measurable disease, and all other patients had at least one mainly lytic bone lesion.All patients had ER-positive tumors. The majority of patients (75%) had received ≥3 previous therapies before the study entry, and 55% of patients had received at least one salvage endocrine therapy and/or salvage chemotherapy.
The median number of treatment cycle was 5.2 (20.8 weeks). Two patients were still on treatment at the data cut-off date (October 17, 2017), with the treatment duration of 22 and 25 months, respectively. Three patients experienced at least one dose reduction of chidamide.

Table 1 Baseline patient characteristics (N=20)
Safety
All 20 patients were assessed for toxicity. The most common AEs of the combination treatment (all grades)were thrombocytopenia (80%), neutropenia (70%), fatigue(55%), leucopenia (50%), vomiting (40%), diarrhea (30%),nausea (25%) and anemia (25%) (Table 2). The treatmentrelated grade 3-4 AEs in more than 2 patients were neutropenia (35%), thrombocytopenia (30%) and leucopenia (20%). Three patients discontinued due to AEs.Four serious adverse events (SAE) were reported in 4 patients. Three SAEs were considered to be related to chidamide, including one grade 3 gastrointestinal dysfunction leading to hospitalization, one grade 2 myelosuppression with poor overall conditions leading to hospitalization, and one grade 3 thrombocytopenia leading to hospitalization. The symptoms resolved after temporary treatment discontinuation or dose reduction of chidamide,and the treatment continued for two patients who had beenhospitalized with myelosuppression. No treatment-related death was reported.
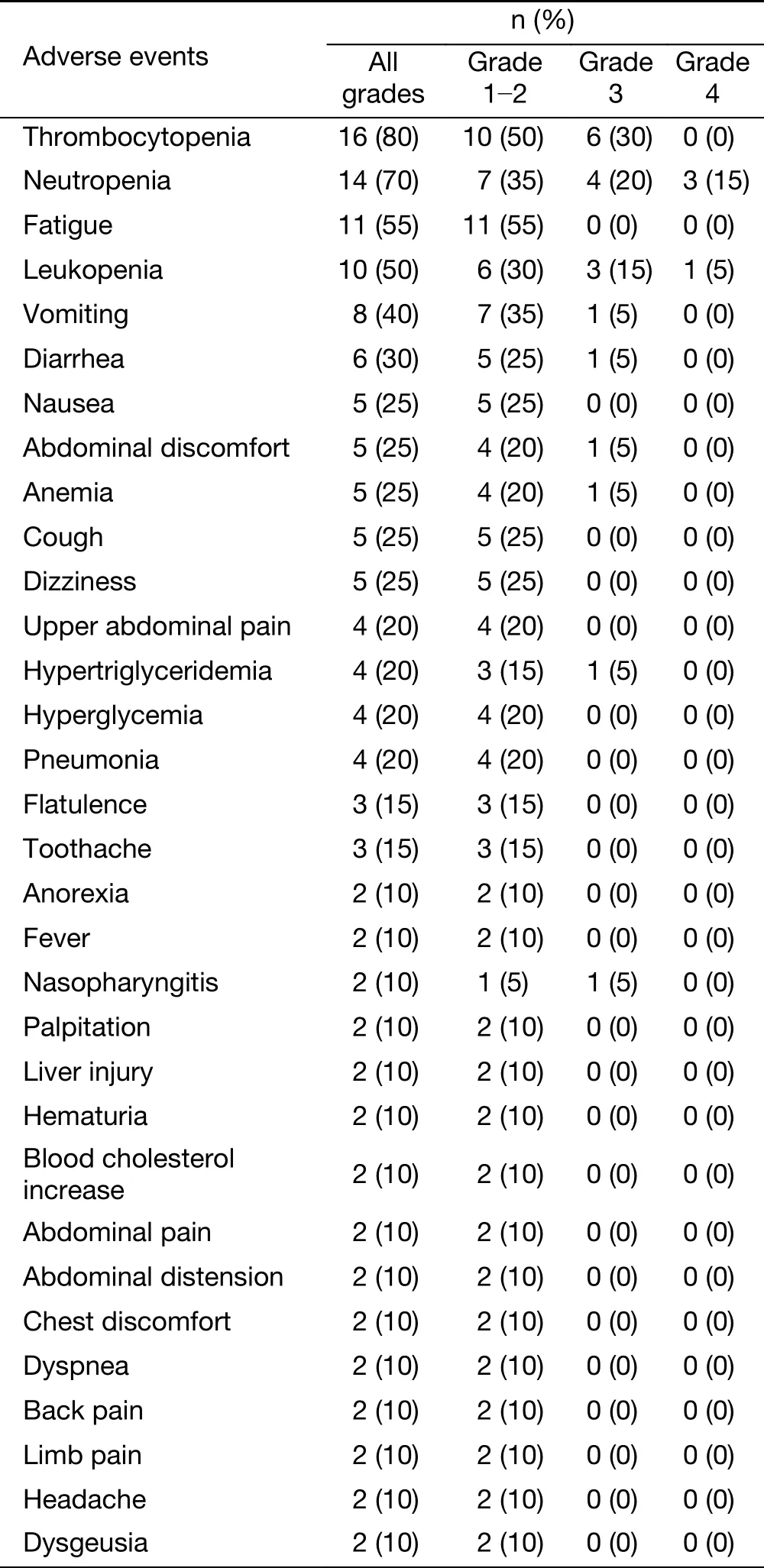
Table 2 Adverse events (occurred in ≥10% patients) (N=20)
PK
Ninteen patients completed the full collection of plasma samples for PK analysis. The PK parameters and the curve of mean plasma concentrations over time of either exemestane or chidamide alone and in combination are shown in Table 3 and Figure 1. There was no significant difference in PK parameters of exemestane before and after combination with chidamide, including the plasma exposure (32.5 vs. 31.4 ng/mL for Cmax, and 71.8 vs. 79.3 ng·h/mL for AUC0-t). For chidamide, while most PK parameters were similar between chidamide alone and combination with exemestane, a higher AUC value was noted after combination with exemestane (1,911.3 vs.2,386.4 ng·h/mL). Inter and intrapatient variations were observed in chidamide PK parameters.
Preliminary efficacy
Best response in 16 of 20 patients (4 patients not evaluable)was assessed by RECIST, including 4 patients with PR, 10 patients with SD, and 2 patients with PD. Of the 14 patients with measurable disease, 4 had PR, 5 had SD, and 2 had PD (Table 4). Median PFS was 7.6 months (231 days)(Figure 2).
Discussion
Primary and secondary drug resistance is one of the major challenges in the treatment of HR+ breast cancer patients with endocrine therapies. Chidamide, functioning as a genuine epigenetic modulator, affects multiple cellular signaling pathways that are abnormally changed in cancer by selective inhibition of HDAC 1, 2, 3 and 10 (13),including the reversal of endocrine resistance in ERpositive breast cancer cells in vitro (17). Several HDAC inhibitors combined with endocrine drugs, including vorinostat and tamoxifen (20), panobinostat and letrozole(21), and entinostat and exemestane (10), have shown the encouraging efficacy in clinical trials in ABC patients. In this exploratory clinical study, we evaluated the safety, PK,and preliminary efficacy of chidamide in combination with exemestane in Chinese patients with HR+ ABC progressed on previous endocrine therapy.

Table 3 PK parameters of exemestane and chidamide

Figure 1 Mean plasma concentration-time curves of exemestane (A) and chidamide (B) alone or in combination treatment.
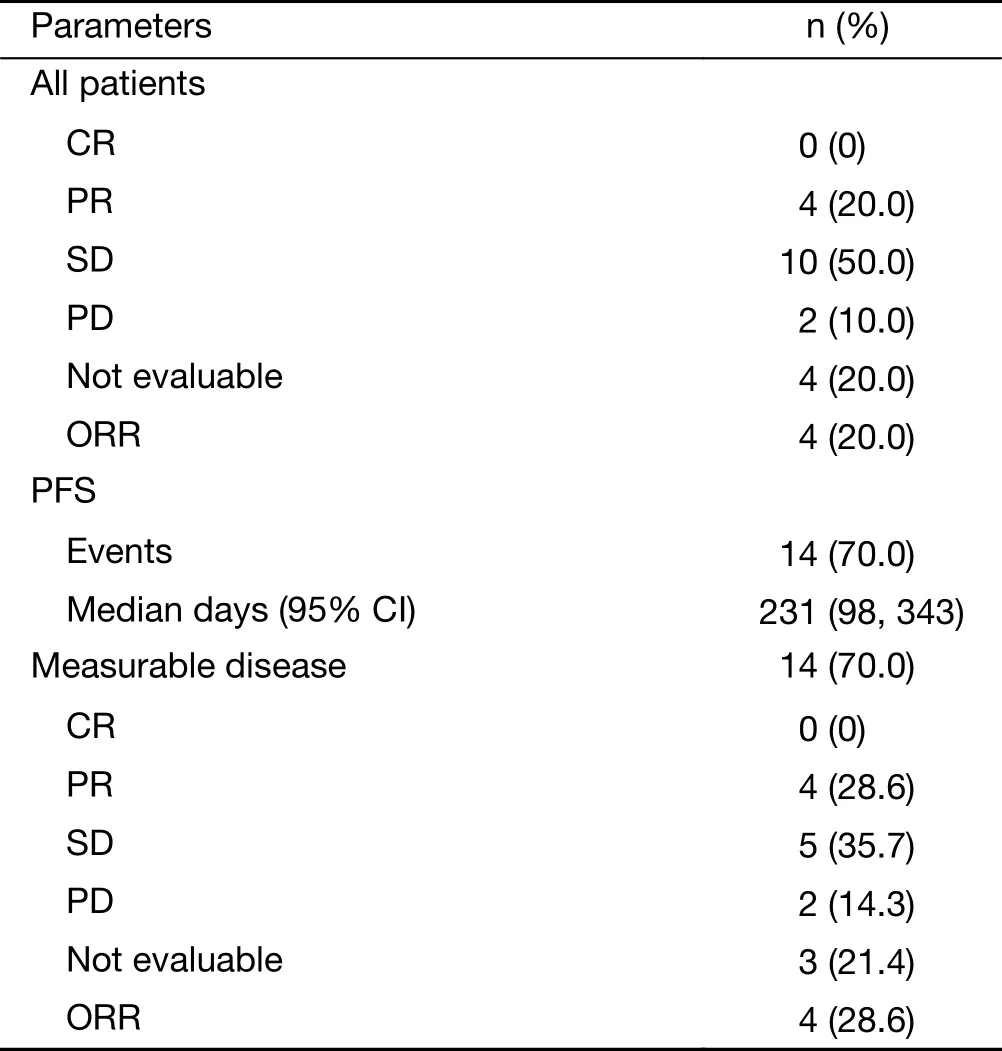
Table 4 Preliminary efficacy (N=20)

Figure 2 Kaplan-Meier estimates of progression-free survival(PFS) probability.
Chidamide 30 mg BIW was the dosing regimen approved for the PTCL indication, which had been shown significant single-agent activity and a manageable safety profile either from the pivotal clinical trial or postmarketing real-world study in relapsed or refractory PTCL patients (12,22). Based on the known PK and safety profiles from each chidamide and exemestane, we employed 30 mg chidamide as the starting dose and kept the opportunity to escalate the chidamide dose down if the potential tolerance and/or drug-drug interaction issues aroused. The results from the study showed that the combination therapy of chidamide 30 mg BIW and exemestane 25 mg daily was generally well tolerated. The toxicity profile was similar to that previously observed from chidamide monotherapy(12,22). The most frequently reported AEs were hematological and gastrointestinal toxicities, including thrombocytopenia, neutropenia, nausea, vomiting and diarrhea. The more frequent grade 3-4 AEs included neutropenia (35%), thrombocytopenia (30%) and leucopenia (20%). The more common hematological AEs were also reported for exemestane monotherapy in ABC patients (23). The apparently higher grade of hematological AEs observed in the current study compared with chidamide-monotherapy in PTCL patients (12), might be due to the overlapping toxicities from each drug in the combination treatment. Most hematological AEs were recovered or relieved after dose reduction or temporary discontinuation of chidamide, and none of the patients withdrew the study due to those AEs.
Previous clinical study of chidamide in combination with paclitaxel and carboplatin in patients with non-small cell lung cancer has shown no evidence that chemotherapy drugs significantly affect PK behaviors of chidamide (19).The overall PK results from this study indicate that combined application of exemestane and chidamide does not affect the PK parameters of each other to a significant degree. While there was little difference in PK behaviors for exemestane before and after combination usage with chidamide, a potential higher blood exposure of chidamide was noted together with exemestane, which was probably due to the inter- and intra-patient variations for chidamide as reported previously (24).
Several exploratory clinical studies have demonstrated promising efficacy results in HR+ ABC patients by combination treatment of HDAC inhibitors and hormone therapies. For example, in a randomized phase II trial,while entinostat in combination with exemestane compared with placebo with exemestane, did not reach PFS primary endpoint (4.3 vs. 2.3 months; P=0.11) and the ORR was similar between the two groups (6.3% vs. 4.6%), the median OS was significantly prolonged in the entinostat group (28.1 vs. 19.8 months; P=0.036) (10). Our results from the current study have shown that combination treatment of ABC patients progressed on previous endocrine therapy with chidamide and exemestane appears to have higher ORR (20%) and longer median PFS (7.6 months) compared with the data from a similar patient population for entinostat + exemestane, as well as exemestane alone (10).
Conclusions
The results of this study show that the combination regimen of chidamide plus exemestane is generally well tolerable with promising preliminary efficacy in HR+ ABC patients progressed on previous endocrine therapy, which encourages further pivotal clinical trial in this patient population.
Acknowledgements
The authors thank all the patients, physicians and nurses who participated in this study. This study was partly supported by grants from the Chinese National Major Project for New Drug Innovation (No. 2015ZX09101005)and the Shenzhen municipal science and technology project (No. JCYJ20120618162903087 and No.JSGG20140515161400837).
Footnote
Conflicts of Interest:The authors have no conflicts of interest to declare.
杂志排行

Chinese Journal of Cancer Research的其它文章
- Liver cancer incidence and mortality in China: Temporal trends and projections to 2030
- Supplementary materials
- Incidence and mortality of lung cancer in China, 2008-2012
- Adjuvant chemotherapy may improve outcome of patients with non-small-cell lung cancer with metastasis of intrapulmonary lymph nodes after systematic dissection of N1 nodes
- Voltage-gated K+ channels promote BT-474 breast cancer cell migration
- Construction and external validation of a nomogram that predicts lymph node metastasis in early gastric cancer patients using preoperative parameters