神经元核内包涵体病1例报道及文献复习
2019-01-17牛延良李建章
牛延良 李建章
1)郑州大学第五附属医院,河南 郑州 450052 2)郑州大学第二附属医院,河南 郑州 450003
1 病例资料
女,74岁,因“头痛、反应迟钝、言语不利1 d”入院。患者1 d前晚餐时突发头痛、恶心,无发热、呕吐,无胸闷胸痛,肢体活动尚可,未在意,少量进食后卧床休息,夜间出现意识模糊,行为异常,胡言乱语,伴大小便失禁,无肢体抽搐,今晨家属发现患者目光呆滞,反应迟钝,言语含糊不清,说话费力,声音低弱,言语交流不能。
既往有原发性高血压史20余年,糖尿病4年余,未规律服药,血糖控制不详。间断头痛数年,长期便秘,间断口服酚酞片。家属述有智能减退2 a,但未曾评估。最近半月随女儿出家居住于山顶寺庙,进食少,每顿饭约1/3碗粥,1/4馒头,基本不吃菜。无有毒有害物质接触史。
入院体格检查:血压131/56 mmHg(1 mmHg=0.133 kPa),神志清,欣快、不时发笑,反应迟钝,理解力差,计算力差,言语低沉无力,双眼球居中,运动无碍,无眼震,双侧瞳孔等大等圆,直径约3 mm,直接及间接对光反射灵敏,右侧鼻唇沟稍浅,构音欠清,咽反射正常,伸舌左偏,肌张力正常,右下肢肌力5-级,余肢体肌力5级,行走稍欠稳,双侧指鼻试验及跟-膝-胫试验欠稳准,闭目难立征不能配合,无不自主运动,双侧巴宾斯基征阴性,四肢腱反射对称性减弱。感觉系统检查不配合。
辅助检查:血同型半胱氨酸41.9 μmol/L,糖化血红蛋白定量6.9%,血糖7.11 mmol/L,抗核抗体滴度1∶80,类风湿全项、电解质、肝肾功能、红细胞沉降率、甲状腺功能、感染四项(梅毒、HIV、乙肝、丙肝)、肿瘤标志物等未见明显异常。心电图未见明显异常。影像学检查见图1。
诊断:可疑神经元核内包涵体病(neuronal intranulear inclusion disease,NIID)。依据老年女性,既往已有智能减退,本次出现意识模糊、行为异常、反应迟钝加重等脑病症状,DWI有特征性的皮髓交界区“火焰状”高信号(图1C、D),T2、FLAIR显示白质内高信号(图1E、F)。但患者拒绝皮肤病理活检。入院后给予前列地尔、神经节苷脂、疏血通等药物治疗,患者意识清,言语不利、反应迟钝较前好转,头痛缓解,家人诉基本恢复至病前状态。目前MMSE 17分。
2 讨论
神经元核内包涵体病(NIID)为一种罕见的以中枢和周围神经系统以及内脏器官嗜酸性透明包涵体为特征的多系统慢性进展性神经变性疾病,多数少年起病,少数中年起病,家族性多见。一般发病10~20 a后死亡。
1968年LINDENBERG在1例精神发育迟缓、进行性痉挛和共济失调的28岁男性患者的脑和内脏细胞内发现了核内包涵体,最先提出了“神经元核内包涵体病(NIID)”的诊断[1]。2011年前欧美报道40余例NIID患者,从婴儿到60余岁不等,多为婴幼儿及青少年,多表现大脑皮质及锥体外系症状,确诊方式主要依靠尸检、直肠活检、神经活检等。直到2011年SONE等[2]学者报道了嗜酸性核内包涵体在皮肤活检标本中的存在,以及DWI显示的特征性皮质-髓质交界高信号病变以来,NIID的诊断主要通过磁共振成像或皮肤活检[3-4]。日本已报道60余例,多为16~68岁起病,主要表现为痴呆、自主神经、周围神经、锥体外系、小脑、精神行为异常等症状,可见散发性和家族性类型,遗传方式未明,曾提出常染色体隐性遗传,但未能确认[5]。通过文献检索目前国内缺乏相关病例报道,2017年温州医科大学附属第一医院陈为安教授、北京天坛医院张在强教授等发起成立了中国NIID协作组群。
本病病理可见弥漫性神经元脱失,中枢神经系统(CNS)、周围神经系统(PNS)、自主神经系统的神经元嗜酸性核内包涵体(neuronal intranuclear inclusion,NII),包涵体是位于核周直径1.5~10 μm圆形物质,泛素阳性、P62阳性,电镜下为无膜结构的纤维物质构成。包涵体主要分布在中枢神经系统(大脑皮质、基底节核、脑干、小脑、脊髓的神经元及胶质细胞)、周围神经(交感神经节、后根神经节、肠管神经丛等);非神经组织-肾小管、皮肤组织(脂肪细胞、纤维母细胞、汗腺细胞)。皮肤活检样本和尸检中,中枢神经系统、皮肤的病理所见是一样的,因此皮肤活检可作为NIID诊断依据。
NIID为异质性疾病,临床症状多变,无特异性,主要表现为中枢神经+周围神经+自主神经症状。(1)中枢神经系统:大脑皮质(持续数小时至数天不等的意识障碍、癫痫、发作性脑病、精神症状等),皮层下(痴呆最常见),锥体外系(震颤、强直),小脑(共济失调);(2)周围神经系统:多发性神经病,远端肌力下降、末梢型感觉障碍;(3)自主神经系统:瞳孔缩小、膀胱功能障碍、晕厥。
根据发病年龄将NIID分为未成年型和成年型。(1)未成年型:NIID常累及青少年,最早可3岁起病,一般在11岁前发病,男女发病相当,一般30岁以前死亡。(2)成年型:成人发病的NIID又分为散发型和家族型。①散发型:发病年龄较晚,平均63.6岁(51~76岁)。痴呆是首发和最主要的临床表现,以认知障碍就诊的占近95%,94%的病人出现瞳孔缩小,50%有运动失调,39%出现意识障碍,33%有膀胱功能障碍,13%有痫性发作。约20%表现为意识障碍、发热、头痛、呕吐等亚急性脑炎样症状。四肢肌力低下、感觉障碍、震颤等症状少见。本病系散发型,主要表现为头痛、反应迟钝、智力减退及精神异常等脑病样症状,为最常见的临床表现。②家族型:根据初发临床症状,分为肌力低下和痴呆两类。肌力下降型平均发病年龄较低,为27.5岁;而痴呆型病人发病年龄与散发型接近,为56.2岁。家族型NIID患者中94%出现四肢肌力降低,72%有感觉障碍,62%有膀胱功能障碍,47.4%有痴呆(其中肌力低下类型9.1%,痴呆类型100%),26%有意识障碍,而痫性发作、脑炎样症状罕见。
影像学检查在本病诊断中具有重要价值,主要表现为DWI在皮髓质交界处(U型纤维)持续性高信号,有称为皮层下火焰征、尿布征,高信号随时间逐渐在皮层扩展加重,不随时间进展而消失,但即使到疾病后期高信号也不会延伸到脑白质,这是NIID特征性影像,只要有这种影像就要疑诊NIID。T2、FLAIR序列出现脑白质高信号,随病情进展出现脑萎缩改变,无特异性;但有学者认为,额叶型白质脑病可能是一个更敏感和早期诊断的指标[6],这与本病例报道相符,白质改变以额叶区最明显。MRI显示小脑萎缩,FLAIR序列小脑蚓部旁及小脑中脚可见高信号,也具有一定特征性,即使使用过去未检查DWI的MR成像检查结果,也可作为诊断NIID的指标[7]。其他在不同序列显示脑萎缩、脑水肿等,智能评分多显示智力差。
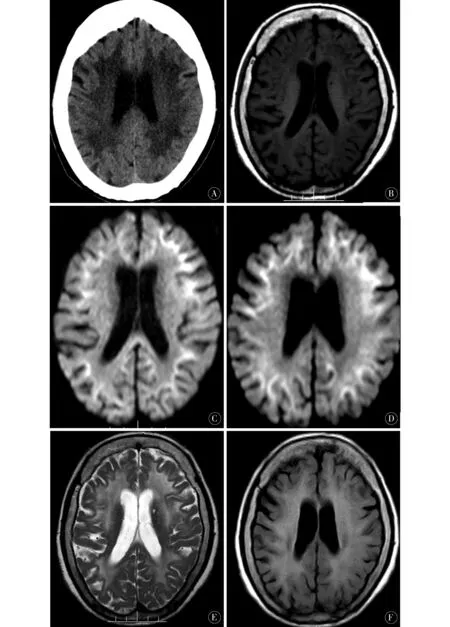
图1 A:CT示脑白质低密度灶;B:T1序列,脑白质低信号;C、D:DWI序列,皮髓质交界处高信号;E、F:分别为T2、FLAIR序列,广泛脑白质高信号Figure 1 A:CT shows white matter low density lesions;B:T1 sequence,white matter low signal;C,D:DWI sequence,high signal at the cortex and medulla junction;E,F:T2,FLAIR sequence,extensive brain white matter high signal
目前NIID无明确统一的诊断标准,依据目前文献资料,以下条件可作为诊断参考标准:(1)临床具有中枢性、周围性、自主性等广泛的神经症状,其中以中枢神经损害症状最重要,为必有条件。(2)影像学以DWI在皮髓质交界处持续性高信号为必有条件,T2、FLAIR序列出现以额叶为主的脑白质高信号为次要参考条件,FLAIR序列小脑蚓部旁及小脑中脚高信号少见,也具有一定特征性。(3)病理检查是诊断金标准,推荐采用皮肤活检,显示嗜酸性核内包涵体。(4)排除其他类似有类似影像表现疾病。符合1、2、3条确诊;符合1、2、4条为可能。本病目前无特异性治疗。
