转录组测序的发展和应用
2019-01-17王楚彪卢万鸿林彦罗建中
王楚彪,卢万鸿,林彦,罗建中
转录组测序的发展和应用
王楚彪1,2,卢万鸿1,林彦1,罗建中1*
(1.国家林业和草原局桉树研究开发中心,广东 湛江 524022;2.南京林业大学,江苏 南京 2100037)
转录组学研究是近年来分子生物学研究的热门,而转录组测序是其核心技术。分子测序技术经历了第一代到第三代的发展,取得长足进步,通量和准确性不断提高,现在应用最广的是二代测序技术,它主要有Roche/454、ABI/Solid、Illumina/Solexa三种测序平台,各有利弊。转录组测序历经基因芯片技术、基因表达系列分析技术、大规模平行测序技术和RNA-Seq技术,目前最活跃的RNA-Seq技术是基于二代测序技术。转录组测序的应用在基因表达水平分析和差异表达分析、新基因的挖掘、寻找单核苷酸多态性及应用、基因功能注释都有所体现。转录组测序和应用是活跃的研究课题,将迅速发展,并在生物研究中起到愈发重要的作用。
转录组;测序技术;高通量;RNA-Seq
在人类基因组计划完成后,进入了探究生物奥秘的后基因时代。基因组学、蛋白质组学和转录组学等逐渐得以应用,由于转录组学研究能相对较快得到结果、容易入手,故迅速发展起来[1]。转录组,通常有广义和狭义之分。广义转录组:指生物体的细胞或组织在一个特定状态下转录出来的所有RNA的总和,包括不编码蛋白质的RNA(包括tRNA,rRNA, micro RNA等)和能编码蛋白质的信使RNA(mRNA)[2];狭义转录组:单指所有信使RNA(mRNA)的总和[3]。转录组的研究有其重要的作用,是基因结构、功能和基因表达的重要研究手段,也是表型关联研究的重要方法。转录组学研究迅猛发展,并已应用在医学、动物、植物等领域应用,其研究的重点是对转录组的测序和分析。转录组学和基因组学比较而言,研究范围更小,针对性更强,因其仅研究被转录的基因[4]。而转录组测序是转录组学研究的关键技术,正是由于转录组测试技术的迅猛发展,推动了转录组学研究进入快车道。
1 转录组测序技术的起步和发展
1.1 高通量测序技术的发展历程
1.1.1 第一代测序技术
DNA测序技术是分子生物学研究的基础和重要技术,它的发展经历了几个重要阶段。早在1975年,研究人员就发明了加减法用于测定DNA序列[5]。两年后,他们对原有测序方法进行改良,引入了双脱氧核苷三磷酸(ddNTP),从而得到了双脱氧链终止法(即Sanger测序法),这很好地提高了DNA序列测定的效率与准确性[6]。同年,MAXAM等[7]报道了通过化学降解以测定DNA序列的方法。以双脱氧链终止法和化学降解法为基础建立起来的DNA测序技术,称为第一代测序技术。第一代测序技术的优点是读长长、精度高,至今仍局部应用于序列的重测序、突变位点的检测等相关研究当中。但是,随着研究的深入和需求的增长,第一代测序方法存在通量小、成本高等方面的缺陷,已经不能满足深度、高通量测序、基因组测序等大规模的测序需求,其应用前景受到了明显的制约[8]。
1.1.2 第二代测序技术
2005年,454生命科学公司(之后被Roche公司收购)首先推出了第二代测序平台Genome Sequencer 20,它是基于焦磷酸测序的,并测定了支原体的基因组序列,打开了第二代测序技术的序幕[9]。很快美国Illumina公司推出了Genome Analyzer测序平台[10],ABI公司推出SOLID测序平台[11],多平台的推出标志着新测序时代的到来。新一代测序技术主要特点是测序时间和成本大幅下降、测序通量大幅提高[12],该技术又称作深度测序技术,是一次革命性变革。该测序技术目前仍然广泛应用于各行业的测序和研究。
边合成边测序(sequencing by synthesis, SBS)是第二代测序技术的中心思想之一,例如Illumina公司的的测序方法,先是将目标DNA打碎成约100 ~ 200个碱基小片段,并在片段两端加上特定的接头序列,构建成为测序文库,之后将要测序的单链的DNA碱基片段利用接头与芯片表面引物进行互补配对,令其一端固定在该芯片上,而另一端和其他引物进行互补固定,构造桥状结构。通过约30轮的扩增反应,每个芯片表面会形成若干亿单克隆DNA簇[8]。接下来,加入4种带有不同颜色荧光标记的dNTP和DNA聚合酶。在DNA合成时,带有荧光标记的核苷酸在引物末端配对时都会释放焦磷酸盐,令荧光标记蛋白放出荧光。之后利用激光扫描反应板来获取各个核苷酸聚合时的荧光颜色,这就能转化为对应的核苷酸序列。重复这个过程,使得每条模板DNA全部聚合为双链。对所有的荧光信号进行统计,可获得各个DNA小片段的序列[8]。
二代测序的3种测序平台各有优缺点,见表1。

表1 不同二代测序平台的比较[12]
注:*成本会有所变化。
Illumina公司的优点是测序性价比最高,其运行成本低,测相同数据量,成本约为454测序的1/10,机器售价也低,缺点是测序片段短。早期的Illumina测序技术只有测序读长20 ~ 30 bp时能保证较高正确率,随着技术的进步,目前高质量的测序读长能达到2 × 150 bp或以上。该公司的Hiseq 4000一次运行能产生的数据量达到150 G。
454 FLX的的优势是测序片段长,能获得读长达400 bp的高质量序列。2008年,该公司全新GS FLX Titanium系列试剂和软件,使测序通量提高了5倍,一次测序可测得万条读长,数据总量约500 M。
SOLID测序的测序读长比较短,然而优点是准确度高,测序数据的准确度大于99.94%,在15 X覆盖率的情况下准确度可达到99.99%,是所有公司测序技术中准确度最高的[13]。
1.1.3 第三代测序技术
二代测序技术通量高,成本低,但存在读长短的问题,使测序后的分析存在不少困难。因此,以单分子测序为特点的第三代测序技术,也开始逐渐进入人们的视野当中。目前主流的三代测序技术有Helico BioScience公司的HeliScope技术[14];Pacific Bioscience公司的SMRT技术[15]等。目前三代测序技术不够完善,因为单分子的荧光信号较弱,单碱基检测的准确率也较低,应用还不广泛。
1.2 转录组测序技术的发展
转录组包括一个细胞的所有转录本信息,是指特定细胞在特定功能状态下全部表达基因的总和,而通常所说的转录组学研究主要是mRNA。转录组测序和分析可以用于发现低丰度转录本、寻找多态性标记、深度挖掘新基因、绘制转录图谱、鉴定基因家族、调控可变剪切、确定代谢途径以及进化分析等研究[16],尤其是分子生物学进入应用阶段的今天,转录组学显得尤为重要。转录组测序的方法有:基因芯片技术(Microarray)[17],基因表达系列分析技术(SAGE)[18],大规模平行测序技术(MPSS)[19]以及RNA测序技术(RNA-Seq)[20]。其中RNA-Seq技术有着高通量、高重复性、宽检测范围、准定量等优点,而且其应用不局限于已知基因组序列信息的物种,对于未知基因组序列的物种也能够使用,是其最大的优势[21]。
1.2.1 基因芯片技术
在“人类基因组计划”进行中,基因芯片技术迅速发展和广泛被应用,是当时功能基因组学研究最重要的研究手段之一。1991年Affymetrix公司在核酸杂交的基础上开发世界上第一块寡核苷酸基因芯片[22]。经过多年的发展,基因芯片技术比较成熟,提高了分析速度,减少了实验所需样品和试剂,实验技术及后期数据分析都是相当成熟,也形成了庞大的公共数据库。缺点一是芯片上探针的信息决定了基因芯片的检测范围,该技术只适用于检测已知序列的情况而没有探索新基因的作用,二是其杂交技术灵敏度不高,很难检测到低丰度基因或捕捉到基因表达水平的细小变化[23]。
1.2.2 基因表达系列分析技术(SAGE)
SAGE技术的技术流程是使用锚定酶切开双链并连接相应的接头,后利用标签酶酶切取得SAGE标签并进行扩增,再将接头序列使用锚定酶切除,获得含标签二聚体的多聚体并对其测序[24]。SAGE技术是以前文提到的Sanger测序为基础的,优点是能很快获得转录图谱。
1.2.3 大规模平行测序技术(MPSS)
对SAGE技术的改进形成了MPSS测序技术。MPSS技术首先将cDNA克隆到具有不同接头的载体库中,再利用PCR扩增载体库中各个cDNA片段,然后利用聚合酶和dGTP的共同作用将PCR产物转换成单链文库,最后通过杂交将其结合在带有Anti-adaptor的微载体上并进行测序。MPSS技术能在较短时间内检测组织或细胞内全部基因的表达情况,在功能基因组研究方面是有效的工具之一[25]。
1.2.4 RNA测序技术(RNA-Seq)
RNA-Seq是近年发展起来也是使用最广泛的转录组测序技术,具有很多优点。一是高分辨率,转录组测序技术可以准确分辨出单个碱基,同时由荧光模拟信号所引起的背景噪音、交叉反应等问题能够有效地避免;二是高通量,通过转录组测序技术不仅能够得到数以亿计个碱基序列,基本能够达到覆盖整个转录组的要求;三是高灵敏度,目标细胞中低至几个拷贝的稀有转录本利用该测序技术也能检测到;四是使用更便捷,该技术能对物种的全转录组进行分析,不需要在测序前设计特异性探针,而是直接分析物种的全转录组[26-28]。
RNA-Seq测序的步骤如下:首先利用纯化的mRNA反转录构建cDNA片段文库,目标mRNA被随机打断并反转录成cDNA或者先进行反转录后再随机打断,之后在文库各片段两端加上测序接头,进行高通量测序。由于测序方法的不同,得到的读长为30 ~ 400 bp。最后,将这些读段比对到参考基因组或转录组上,目的是进行拼接,或者直接计算转录本的一些参数,例如表达量;如果没有参考基因组,则进行de novo拼接,之后再进行计算相关参数,而要对转录组进行更深入的研究,则需要借助其他技术,例如数字基因表达谱技术[21],其流程可参考图1。
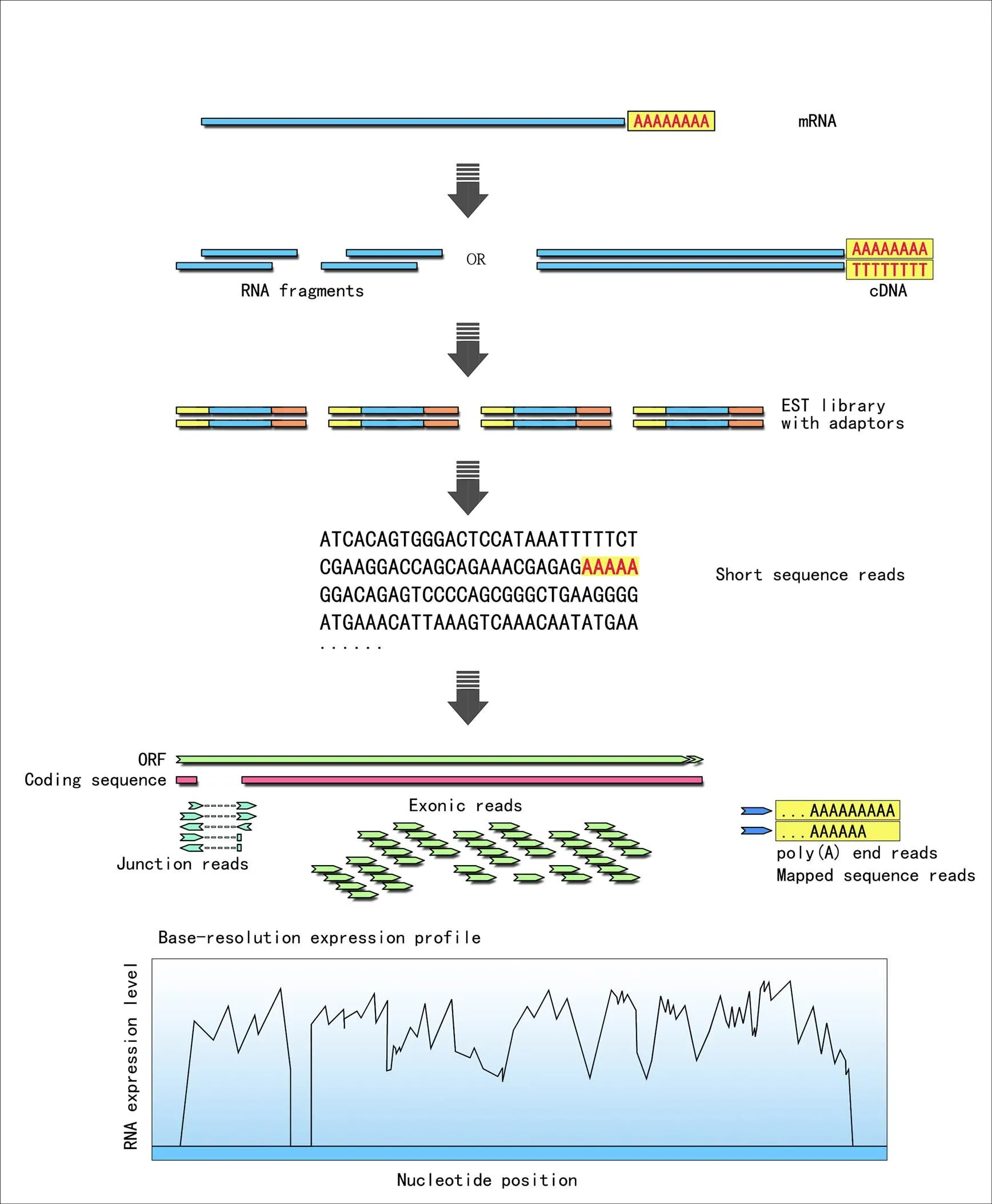
图1 RNA测序及分析的典型流程[29]
RNA-Seq是二代测序技术的一个重要应用,近来发展较为迅速,已成为对生物体进行转录组分析和基因表达定量分析的重要途径[30]。利用RNA-Seq对生物体进行转录组测序分析,可以补充扩展该物种的基因数据库,获得大量的相关ESTs信息,发掘一些新的功能基因,有利于后续的基因克隆和相 关分子标记的开发,还可以研究特定组织或细胞基因的时空表达和探索一些未知的小RNA等,为后 续的研究与应用提供理论基础[31]。
2 转录组测序的应用
2.1 基因表达水平分析和差异表达分析
生物体细胞中基因的表达特性可以通过mRNA水平(浓度)的测量来表示,在任何组织中以不同水平进行表达均可检测。由于存在转录后水平调控(干扰RNA),相关的mRNA和相关的蛋白之间的联系并不一定强烈,但是测量mRNA的浓度依然是检测细胞相关表达水平和健康与否的一个重要指标[32]。唯一能够准确判断个体基因发生突变的方法是与种系的转录组序列进行比较。而表达谱芯片技术可用于研究个体、时间、基因对表达的影响,即相同个体在同一时间不同基因的表达差异,相同个体在不同时间里相同基因的表达差异;不同个体的在相同时间相同基因的表达差异等,主要体现表达量的不同[33]。
RNA差异表达分析主要是细胞在不同情况下的表达差异。RNA测序能够检测整个转录组的能力,使得它成为检测生物体基因表达的重要工具。生物信息学家发明专用自动化系统来管理数据数量庞大的序列,创造新的算法和软件进行测序结果的比较。RNA-Seq 数据库已经被用来寻找在特殊途径中的基因[34]。RNA-Seq数据的在微列阵平台分析的主要优点是可以覆盖整个转录组,从而有可能解开基因调控网络,也可以用于检测和预测与它们的生物学功能相同的基因的剪接。
2.2 新基因的挖掘
转录组测序一般是对生物体可表达的全部基因的测序,将得到的序列与公共数据库中已知的序列进行比对,则可以寻找出新的基因并大致预测其功能[35],甚至不同物种间的比对也能够挖掘出目标物种的一些基因。在高等植物基因组测序之后能够对该物种的基因组进行组装和拼接,并且对基因进行QTL定位和功能预注释分析,但是目前的研究水平对很多基因研究不深入,位置把握不准,这时候就需要通过转录组测序,并与物种的性状进行关联分析,从而对目标基因的分析进行优化。
2.3 单核苷酸多态性分析
转录组测序之后通过比对到参考基因组能够发现大量的SNP (single nucleotide polymorphism),对SNP的深入分析对生物学的研究具有重要意义。早期转录组单核苷酸发掘能够在Roche 454 sequencing平台进行分析,而在进行sanger sequencing 验证中,研究人员能够在 2 400多个玉米基因获得差不多5 000个保守的单核苷酸多态性[36]。随着测序技术的发展,能够发现的SNP数量越来越多,转录组测序已成为研究生物环境的影响、发育调控、细胞类型等较复杂分子机制的重要手段,同时也是应用于SSR和SNP等分子标记多态性鉴定的重要前提[37]。RAJEEV等[38]对292个木豆属()种质进行测序,共取得了1 510万个SNP,其中在基因区域的SNP达到302万,对SNP的分析并与性状进行关联,得到了木豆种质的差异和木豆相关性状的关联基因区域。
2.4 基因功能注释
对转录组进行测序之后能较直接地进行基因功能注释。基因功能注释需要利用生物信息学方法,将测序得到的未知基因序列在公共数据库进行比对,通过分析与公共数据库中已知基因的聚类或同源性,来预测目标未知基因的功能。目前使用的基因功能预测分类系统主要是Gene Ontology(GO)分类和KEGG功能分类[39]。GO采用的思想是聚类分析,聚类是将同一组中的对象与相似的其他组(簇)相比较,从而推测出目标基因的功能。聚类分析包括层次聚类、K-均值聚类、K-中心点聚类和基于网络或模型等的一些聚类技术[40]。KEGG是基于分子水平信息,特别是大型分子数据集合而生成的基因组测序数据库和其他高通量实验得出的数据库资源,是一个有关Pathway的主要公共数据库,在给出一套完整基因的情况下,它可以对蛋白质在各种细胞活动中的作用作出预估[41]。
3 展望
由于转录组是参与表达的基因组合,对生物体各性状的表现具有非常重要的作用,也是基因功能关系最密切的组学,所以目前转录组测序是分子生物学发展最迅速,应用相对最广的一种测序形式,几乎所有常见并在研的生物体都有进行转录组方面的研究。转录组学的研究方向不断拓展,在自然群体和遗传群体的材料中有基因定位、基因功能注释、遗传进化分析和比较转录组学等方面的分析,在个体材料中主要有发育调控、环境适应,、表观调控等方面的分析。在大量生物体进行转录组研究的背景下,越来越多的基因被发现,功能被注释,对生物体的研究将愈发深入,而基因功能有一定的共通性,这也使得相近物种的研究更加容易。
测序技术日新月异,随着三代测序技术的不断完善,其实际应用也更加临近,从而使转录组测序的结果更加准确完整,未来转录组测序在生物学研究中将扮演更加重要的角色,转录组测序应用将更广,成本更低,使用也更加便捷高效。
[1] LOCKHART D J, WINZELER E A. Genomics, gene express and DNA arrays[J].Nature,2000,405(6788):827-836.
[2] COSTA V, ANGELINI C, DE FEIS, et al. Uncovering the complexity of transcriptomes with RNA-Seq[J]. Jorunal of Biomedicine and Biotechnology, 2015, 2010(5757): DOI:10.1155/2010/853916.
[3] 张春兰.小尾寒羊和杜泊羊臂二头肌转录组及肌球蛋白轻链基因家族结构特征分析[D].泰安:山东农业大学,2014.
[4] ANAORGE W J. Next-generation DNA sequencing techniques [J]. New Biotechnology, 2009, 25(4): 195-203.
[5] SANGER F, COULSON A R. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase[J]. Journal Molecular Biology,1975,94(3): 441–448.
[6] SANGER F, NICKLEN S, COULSON A R. DNA sequencing with chain-terminating inhibitors. Proceedings of the National Academy of Sciences[J]. 1977,74(12):5463-5467.
[7] MAXAM A M, GILBERT W. A new method for sequencing DNA[J]. Proceedings of the National Academy of Sciences, 1977, 74(2):560-564.
[8] 解增言,林俊华,谭军,等.DNA测序技术的发展历史与最新进展[J].生物技术通报,2010(8):64-70.
[9] MARGULIES M, EGHOLM M, ALTMAN W E, et al. Genome sequencing in microfabricated high-density picolitre reactors[J]. Nature, 2005, 437(7057): 376-380.
[10] PORRECA G J, ZHANG K, LI J B, et al. Multiplex amplification of large sets of human exons[J]. Nature Methods,2007,4(11):931-936.
[11] ONDOV B D, VARADARAJAN A, PASSALACQUA K D, et al. Efficient mapping of Applied Biosystems SOLiD sequence data to a reference genome for functional genomic applications[J]. Bioinformatics,2008,24(23):2776-2777.
[12] SHENDURE J, JI H. Next-generation DNA sequencing[J]. Nature Biotechnology, 2008,26(10):1135-1145.
[13] 陈浩东.达尔文氏棉旱胁迫转录组测序、EST-SSR开发及高密度遗传图谱构建[D].北京:中国农业科学院,2013.
[14] 王丽鸳.基于EST数据库和转录组测序的茶树DNA分子标记开发与应用研究[D].北京:中国农业科学院,2011.
[15] HARRIS T D, BUZBBY P R, BABCOCK H, et al. Single-molecule DNA sequencing of a viral genome[J]. Science, 2008, 320(5872): 106-109.
[16] EID J, FEHR A, GRAY J, et al. Real-time DNA sequencing from single polymerase molecules[J]. Science,2009, 23(5910): 133-138.
[17] 侯婷婷.缺血再灌注脊髓损伤不同时序变化转录组学相关研究[D].长春:吉林大学,2015.
[18] DUGGAN D J, BITTNER M, CHEN Y, et al. Expression profiling using cDNA microarrays[J]. Nature Genetics, 1999, 21(1):10-14.
[19] VELCULESCU V E, ZHANG L, VOGELETEIN B, et al. Serial analysis of gene expression[J]. Science, 1995, 270(5235) :484-487.
[20] HENE L, SREENU V B, VUONG M T, et al. Deep analysis of cellular transcriptomes-Long SAGE versus classic MPSS[J]. BMC Genomics, 2007(8): DOI: 10.1186/1471-2164-8-333.
[21] BRAUTIGAM A, GOWIK U. What can next generation sequencing do for you? Next generation sequencing as valuable tool in plant research [J]. Plant Biology, 2010, 12(6): 831-841.
[22] 王少甲.基于转录组测序的小金海棠缺铁胁迫相关基因研究[D].北京:中国农业大学, 2014.
[23] 郭溆.基于转录组测序的石斛生物碱和人参皂苷生物合成相关基因的发掘、克隆及鉴定[D].北京:北京协和医学院,2013.
[24] MA Y, YUAN L,WU B, et al. Genome-wide identification and characterization of novel genes involved in terpenoid biosynthesis in Salvia miltiorrhiza[J]. Journal of Experimental Botany, 2012, 63 (7): 2809-2823.
[25] ANDREW J S, GREGORY G, NICOLAS P, et al. Peroxisomal localisation of the final steps of the mevalonic acid pathway in[J]. Planta, 2011, 234 (5): 903-914.
[26] HU Y, WALKER S. Remarkable structural similarities between diverse glycosyltransferases [J]. Chemistry and Biology, 2002, 9 (12): 1287-1296.
[27] WILHELM B T, MARGUERAT S, WATT S et al. Dynamic repertoire of a eukaryotic transcriptome surveyed at single nucleotide resolution[J]. Nature, 453(7199): 1239-1243
[28] 梁烨,陈双燕,刘公社.新一代测序技术在植物转录组研究中的应用[J].遗传,2011,33(12):1317-1326.
[29] 贾昌路,张瑶,朱玲,等.转录组测序技术在生物测序中的应用研究进展[J].分子植物育种,2015,13(10):2388-2394.
[30] ZHONG W, MARK G, MICHAEL S. RNA-Seq: a revolutionary tool for transcriptomics [J]. Nature Reviews Genetics, 2009, 10(1):57-63.
[31] AUFFARY C, HOOD L. Editorial: Systems biology and personalized medicine-the future is now [J]. Biotechnology Journal , 2012, 7(8) : 938–939.
[32] XIANG Z, ZHAO P, LI Q, et al. MicroRNAs ofidentified by Solexa sequencing [J]. BmcGenomics,2010(11):DOI:10.1186/1471-2164-11-148.
[33] GREENBAUM D, COLANGELO C,WILLIAMS K, et al. Comparing protein abundance and m RNA expression levels on a genomic scale [J]. Genome Biology, 2003, 4(9): DOI:10.1186/gb-2003-4-9-117.
[34] Li H, LOVCI M T, KWON Y, et al. Determination of tag density required for digital transcriptome analysis: application to anandrogensensitive prostate cancer model [J]. Proceedings of the National Academy of Sciences, 2008,105(51): 20179-20184.
[35] MARCOTTE E M, PELLEGRINI M, THOMPSON M J ,et al. A combined algorithm for genomewide prediction of protein function [J]. Nature,1999, 402(6757): 83-86.
[36] 郝大程,马培,穆军,等.中药植物虎杖根的高通量转录组测序及转录组特性分析[J].中国科学,2012,42(5):398-412,431-433.
[37] BARBAZUKK W B, SCOTT J E, HSIN D C, et al. SNP discovery via 454 transcriptome sequencing [J]. The plant journal, 2007,51(5): 910-918.
[38] 刘峰,谢玲玲,弭宝彬,等.辣椒转录组SNP挖掘及多态性分析[J].园艺学报,2014,41(2):343-348.
[39] RAJEEV K V,RACHUIT K S,HARI D U, et al. Whole-genome resequencing of 292 pigeonpea accessions identifies genomic regions associated with domestication and agronomic traits[J]. Nature Genetics.2017,49(7):1082–1088.
[40] 黄小花,许锋,程华,等.转录组测序在高等植物中的研究进展[J].黄冈师范学院学报,2014,34(6):28-35.
[41] ESTIVILLCASTRO V. Why so many clustering algorithms: a position paper [J]. Acm Sigkdd Explorations Newsletter,2002,4(1): 65-75.
[42] KANEHISA M. et al., KEGG for representation and analysis of molecular networks involving diseasesand drugs[J].Nucleic acids research,2010,38(suppl 1):355-360.
Development and Application of Transcriptome Sequencing
WANG Chubiao1,2, LU Wanhong1, LIN Yan1, LUO Jianzhong1
(1.,,,; 2.)
Transcriptomics has become a hot topic in molecular biology research in recent years. Transcriptome sequencing, which has emerged as a core technology for molecular genetics research, has progressed rapidly from its first to third generation methodologies with great improvement in throughput and accuracy. Currently, the most widely used is second generation sequencing methodology employing one of three sequencing platforms: Roche/454, ABI/Solid, and Illumina/Solexa, each of which has advantages and disadvantages. All of these methodologies rely on transcriptome sequencing using gene chips and examination of gene expression using SAGE, MPSS or RNA-Seq methods, with the latter being that most commonly used. Applications of transcriptome sequencing include analyses of gene expression levels and differential gene expression, mining for new genes, SNP discovery and annotation of gene functions. Transcriptome sequencing is forecast to continue to be an active research area that will continue to develop rapidly and play an increasingly important role in biological research.
Transcriptome; sequencing technology; high throughput; RNA-Seq
Q752
A
广东省林业科技创新项目(2017KJCX031, 2018KJCX027)。
王楚彪(1982— ),男,在读博士,助理研究员,主要从事林木遗传育种研究,E-mail:scauwcb@163.com.
罗建中(1969— ),男,博士,研究员,硕导,主要从事林木遗传育种研究,E-mail:luojz69@hotmail.com.
