谷子分蘖相关QTL定位及紧密连锁标记开发
2019-01-09杜晓芬李云飞王智兰袁国保杜国华彭建祥张文娜崔巨多郭二虎邹洪锋张林义彭书忠
杜晓芬,王 军,李云飞,王智兰,袁国保,杜国华,韩 芳,彭建祥,张文娜,蔡 伟,袁 峰,崔巨多,郭二虎,邹洪锋,张林义,彭书忠
(1.山西省农业科学院 谷子研究所,山西省特色杂粮种质资源发掘与育种重点实验室,山西 长治 046011;2.华大农业应用研究院,广东 深圳 518083;3.华大小米产业股份有限公司,广东 深圳 518083;4.延安市农业科学研究所,陕西 延安 716000)
对于许多主要农作物(如小麦、水稻、大麦和高粱等)来说,分蘖是与产量直接相关的重要农艺性状之一[1]。一方面,分蘖会影响整个植株的株型,导致植株的光吸收能力、开花和结实率发生变化,最终使产量发生改变[2];另一方面,分蘖可以产生不定根,这使得分蘖株能够在没有主茎的情况下正常生长,从而维持一定的群体产量[1,3]。
分蘖是由主茎基部的节发生的分枝,其形成是许多因子相互作用的结果,这些因子包括环境信号、激素途径和基因调控网络[4-5]。前人已经对一些主要禾谷类作物的分蘖遗传机制进行了广泛研究。水稻的研究结果表明,光、温、水、肥等环境因素[6-7],生长素、细胞分裂素、独脚金内酯[8]等激素都会影响水稻分蘖的形成和生长发育。同时,水稻中相继克隆了11个控制分蘖的基因,包括水稻分蘖过程中的关键调控基因MOC1[9],其余几个为多蘖矮秆基因(D3、D17、D10等)[10-18],这些基因与激素共同调控分蘖形成的相关网络也得到解析[19-20]。除此之外,分蘖通常还受数量性状位点(QTL)控制。根据Gramene网站(http://archive.gramene.org/qtl)统计结果,目前水稻在不同群体中已经鉴定分蘖相关QTL共计213个,高粱中共计19个。小麦中也有一些控制分蘖主效位点的研究报道[21-23]。近年来,谷子中分蘖相关QTL的定位研究也有相继报道。Doust等[24-25]利用F2∶3群体分别在不同温度和不同密度种植条件下共鉴定了41个谷子分蘖相关QTL;Jia等[26]利用全基因组关联方法鉴定了5个环境条件下共计57个与分蘖相关的位点;Mauro-Herrera等[3]利用重组自交系群体分别在温室和大田条件下,检测到谷子苗期、开花期和成熟期共42个控制分蘖相关QTL;Zhang等[27]利用重组自交系群体分别在长日照和短日照条件下共检测到6个控制分蘖相关QTL。尽管如此,有关环境因素和激素因子如何影响谷子分蘖的形成和生长发育,其机制还不清楚。
与其他主要农作物相比,谷子农艺性状基因或QTL发掘研究还比较落后。随着谷子参考基因组的公布[28-29],谷子中许多遗传研究相继开展,尤其是多种类型的分子标记被开发并应用于基因精细定位、基因克隆和分子标记辅助选择。Sato等[30]和Masumoto等[31]利用SSR(Simple sequence repeat)和TD(Transposon display)标记完成了谷子stb1和NEKODE1基因的精细定位;利用SNP(Single nucleotide polymorphism)、CAPS(Cleaved-amplified polymorphic sequence)和InDel(Insertion-deletion)等不同分子标记,谷子SiDWARF2、SiDWARF3、SiYGL1、SiAGO1b和Loosepanicle1等基因相继精细定位和克隆[26, 32-37]。随着下一代测序技术的发展,涌现出了不少新的快速对目标基因或QTL进行定位的方法,其中包括简化基因组测序技术-RAD-seq,该技术利用限制性内切酶对基因组进行酶切,产生一定大小的片段,构建测序文库,对酶切后产生的RAD标记进行高通量测序,具有通量高、准确性高、性价比高、试验周期短和不受基因组序列限制等优点[38],已经广泛应用于SNP标记开发、高密度遗传图谱构建、植物重要性状的QTL定位等研究领域。谷子中也有利用RAD-seq技术对株高、米色等重要农艺性状进行QTL定位的研究报道[39-40]。
本研究采用简化基因组测序技术(RAD-seq),以2个谷子分蘖型农家种(母本)和2个不分蘖育成种(父本),为亲本杂交获得F1,F1各自自交后获得的2个F2群体为主要研究对象, 对谷子分蘖相关QTL进行了分析;同时,新开发了与分蘖QTL(qAJTN7-3和qHCTN7)紧密连锁的分子标记,旨在为谷子分蘖基因的克隆和解析分蘖遗传机制奠定基础。
1 材料和方法
1.1 试验材料
本研究分别以2个F2分离群体为主要研究对象,其中,矮宁黄×晋谷21号杂交组合,以分蘖农家种矮宁黄为母本、不分蘖育成种晋谷21号为父本,命名为Cross AJ;红苗粘谷×长农35号杂交组合,以分蘖农家种红苗粘谷为母本、不分蘖育成种长农35号为父本,命名为Cross HC(图1)。于2014年获得2个杂交组合的F1单株,2015年将收获的F1单株及其亲本种植于山西省农业科学院谷子研究所试验田,通过2个杂交组合F1单株分别自交,获得了2个不同的F2遗传群体(Cross AJ、Cross HC)。为便于调查,亲本及群体均采用宽窄行种植模式,宽行行距0.48 m,窄行行距0.18 m,行长3 m。于谷子分蘖期调查分蘖数(含主茎),亲本取5株调查,取其平均数作为分蘖数值,F2群体逐株挂牌调查。另外,在本研究中,用于验证InDel标记和分蘖数间相关性的160份谷子种质(部分种质由中国农业科学院国家农作物种质资源库提供)的种植模式、调查方式与F2群体及其亲本相同。
1.2 试验方法
1.2.1 DNA样品准备和提取 DNA取样:待幼苗长至三叶一心时,剪取适量叶片放入2 mL离心管内,然后迅速放入-80 ℃超低温冰箱保存备用。谷子基因组DNA提取采用CTAB法[41]。
1.2.2 连锁图谱构建和QTL分析 RAD-seq试验方法及步骤参照Baird等[42]描述进行,参考基因组版本Setaria_italica_v2.0(https://www.ncbi.nlm.nih.gov/genome/?term=foxtail+millet),选取限制性内切酶EcoRⅠ(Cross AJ)和TaqⅠ(Cross HC)分别酶切基因组DNA用于构建RAD-seq DNA文库,使用Illumina HiSeq X Ten平台进行测序。根据识别标签序列得到每个个体的测序reads,使用BWA(Burrows-Wheeler Aligner)[43]软件将个体reads与参考基因组进行序列比对。比对结果进行格式转换后使用GATK(Genome Analysis ToolKit)[44]软件和SAMtools[45]软件用于检测SNP和过滤SNP,SNP鉴定和基因分型方法参照Wang等[39]描述。使用MSTmap[46]软件用于分子标记排序(主要参数:作图函数为Haldane、P值为0.000 000 1、缺失阈值为0.3、目标函数为最大似然法)和连锁图谱构建(取最小阈值LOD 5.0对所有标记进行分组,每个连锁群上标记顺序通过两两标记之间的最小重组频率计算)。
利用WinQTLCart 2.5[47]软件中复合区间作图法(CIM)进行分蘖数QTL定位。进行CIM分析时,选用步长(Walking speed)1 cM和滑窗(Scanning window)10 cM,按照模型6,选取1 000次排列测验,显著水平设置为0.05,判断是否存在QTL。QTL的命名方法为:q加群体简称(AJ、HC),后面加性状简写(TN),最后加染色体名称(当同一条染色有多个QTL时加-1,-2)。
1.2.3 分子标记开发 本研究利用GATK[44]软件搜索InDel,InDel长度设定在1~10 bp。根据选定InDel位点的侧翼序列(上游250 bp,下游250 bp)设计引物进行验证,引物设计采用Primer 3.0(http://primer3.ut.ee/)在线设计,引物由天一辉远(北京)有限公司合成,本研究用到引物序列为MRI5F:5′-GTAGTCGTCCGTCCAAGC-3′,MRI5R:5′-CACCAC CATCAAACAAAGG-3′;MRI322F:5′-GGCAATCCCAA GTATGTGCA-3′,MRI322R:5′-AAGGGAGGAAGTGAA GGTGG-3′;MRI348F:5′-TCTGGTCACCTGTCGTTCTC-3′,MRI348R:5′-AAGTAAGCCACCGTAGCCTT-3′;MRI351F:5′-AAGTTGCCCTTAACCCACCT-3′,MRI351R:5′-GACATTGCCTCGCCGTAAAA-3′;MRI381F:5′-AACC TCCACCATGAAACCCT-3′,MRI381R:5′-CTGGGAGG AAAGAGGGAGTG-3′。
PCR反应体系为10 μL,包括:1 μL 10×PCR Buffer,1 μL 双向引物(2 μmol/L),0.8 μL dNTP(2.5 mmol/L),0.1 μLTaqDNA聚合酶,1 μL 模板DNA(50 ng/μL),灭菌水 6.1 μL。PCR反应程序为:94 ℃预变性5 min;94 ℃变性30 s,58 ℃退火30 s,72 ℃延伸30 s,35个循环;最后72 ℃终延伸5 min。
PCR扩增产物在8%非变性聚丙烯酰胺凝胶进行电泳,根据Marklund等[48]描述方法银染显色,拍照后统计存档。
1.3 数据分析
亲本和F2群体的表型分析、分子标记分型与160份谷子种质(包括112不分蘖种质和48份分蘖种质)表型相关性分析采用统计分析软件SPSS 17.0。
2 结果与分析
2.1 表型分析
矮宁黄×晋谷21号群体(Cross AJ)共获得543个F2单株,红苗粘谷×长农35号群体(Cross HC)共获得131个F2单株。亲本表型如图1所示,2个F2群体及其亲本分蘖数统计如表1所示。对于Cross AJ,亲本之间有较大差异,其F2群体变异范围为1~9;对于Cross HC,亲本差异较Cross AJ群体的亲本略小,F2群体变异范围为1~4。

表1 两个F2群体及其亲本分蘖数分析Tab.1 Phenotypic data of tiller number in two F2 populations

图1 两个F2群体亲本的表型Fig.1 The phenotype of four parents in two F2 populations
2.2 基于RAD-seq的高密度遗传图谱的构建
本研究利用RAD-seq技术,对Cross AJ的亲本和543个F2单株、Cross HC的亲本和131个F2单株进行测序。结果表明,Cross AJ共获得88.33 Gb的原始数据,对原始数据初步分析后,统计出数据的平均测序深度为4.55×,平均上图率为89.77%;Cross HC共获得872 Mb的原始数据,平均测序深度3.12×,平均上图率89.37%(表2)。
通过SNP鉴定后,筛选亲本间可信度高的SNP多态性标记,然后对群体个体进行基因型检测,最后利用MSTmap软件绘制遗传连锁图谱。Cross AJ共获得48 790个上图SNP标记,总图距为1 461 cM;Cross HC共获得9 968个上图SNP标记,总图距为1 648.8 cM(表2)。

表2 两个F2群体的RAD-seq统计结果Tab.2 Summary of RAD-seq in two F2 populations
2.3 分蘖相关QTL分析
结合2个F2群体的表型数据和SNP标记分型结果,利用WinQTLCart 2.5软件进行分蘖相关QTL定位。本研究共鉴定出8个QTL(表3)。在Cross AJ群体中,共鉴定了6个QTL,分别为qAJTN1、qAJTN5、qAJTN7-1、qAJTN7-2、qAJTN7-3和qAJTN9,解释表型变异0.7%~9.8%;在Cross HC群体中,共鉴定了2个QTL,分别为qHCTN5和qHCTN7,解释表型变异1.4%~8.3%。在这些QTL中,效应较大的有qAJTN5、qAJTN7-3和qHCTN7,分别解释表型变异的9.8%,9.3%和8.3%,其加性效应值分别为0.58,0.45和0.43,说明其增效等位基因来自母本矮宁黄或红苗粘谷。
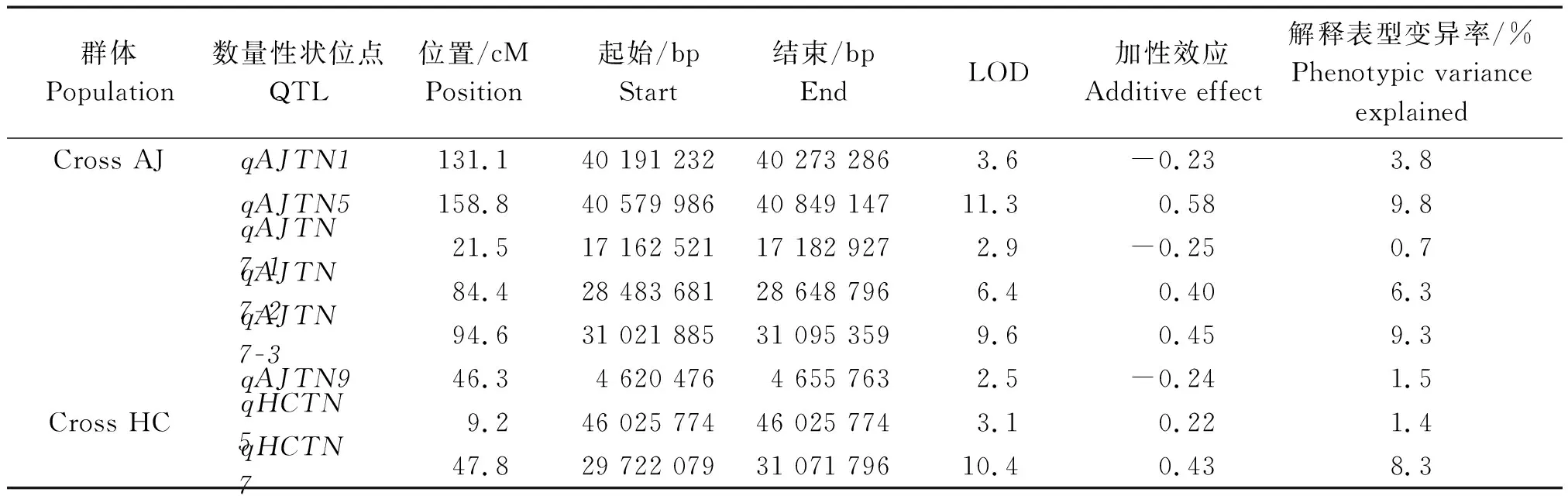
表3 分蘖相关QTL分析Tab.3 QTL analysis of tillering in two F2 populations of foxtail millet
2.4 分蘖数紧密连锁InDel标记开发
在2个群体中各选择了1个效应较大的QTL(qAJTN7-3和qHCTN7),并在其定位区间内开发了新的紧密连锁分子标记,以有利于分蘖性状的分子标记辅助选择。首先,将亲本基因组测序结果与豫谷1号参考基因组进行序列比对,搜索插入缺失位点,设计引物进行PCR扩增,扩增片段进行聚丙烯酰胺凝胶电泳验证,筛选在亲本之间具有多态性的标记,进一步获得在F2后代中能够良好分型的InDel标记(图2-A)。其次,利用MSTmap软件对新标记进行遗传连锁分析。连锁分析结果发现,标记MRI351和MRI322定位在qAJTN7-3区间内;MRI351、MRI5、MRI348和MRI381定位在qHCTN7区间内(图2-B)。
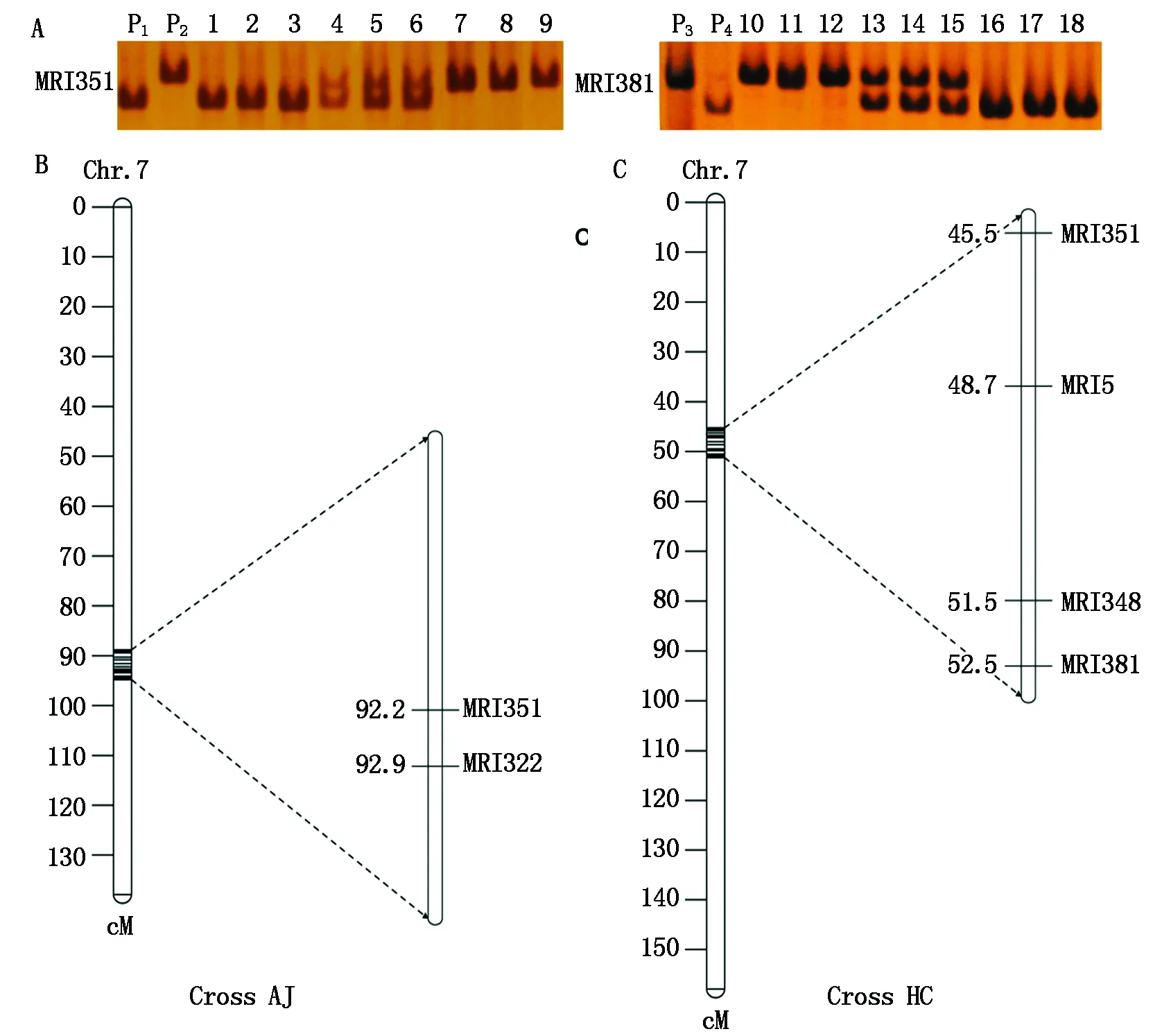
A.标记MRI351和MRI381在亲本和2个F2群体部分单株中的聚丙烯酰胺凝胶电泳结果;B、C.标记MRI5、MRI348、MRI322、MRI351和MRI381在各自染色体上的位置。P1.矮宁黄,P2.晋谷21,P3.红苗粘谷,P4.长农35;1-9为Cross AJ的部分F2单株,其中,1-3单株表型为分蘖,4-9表型为不分蘖;10-18为Cross HC的部分F2单株,其中,10-12单株表型为分蘖,13-18表型为不分蘖。
为了进一步确认InDel标记和分蘖数间的相关性,选取了2个F2群体中的标记MRI351和MRI381,对160份谷子种质进行了基因分型和相关性分析(表4)。根据分子标记检测结果,将与分蘖亲本带型一致的基因型标记为“1”,与不分蘖亲本带型一致的基因型标记为“0”,与双亲带型均不一致的基因型标记为“2”,没有扩增结果的标记为“-”。利用统计分析软件SPSS 17.0的Pearson′s双侧检验,得到相关系数R分别为0.280(标记MRI351)和0.242(标记MRI381),且差异达到极显著水平(表5)。上述结果表明,MRI351和MRI381与分蘖性状紧密连锁,可应用于分蘖型谷子分子标记辅助育种。

表4 160份谷子种质和标记MRI351、MRI381基因分型结果Tab.4 The genotype of MRI351 and MRI381 in the 160 foxtail millet accessions
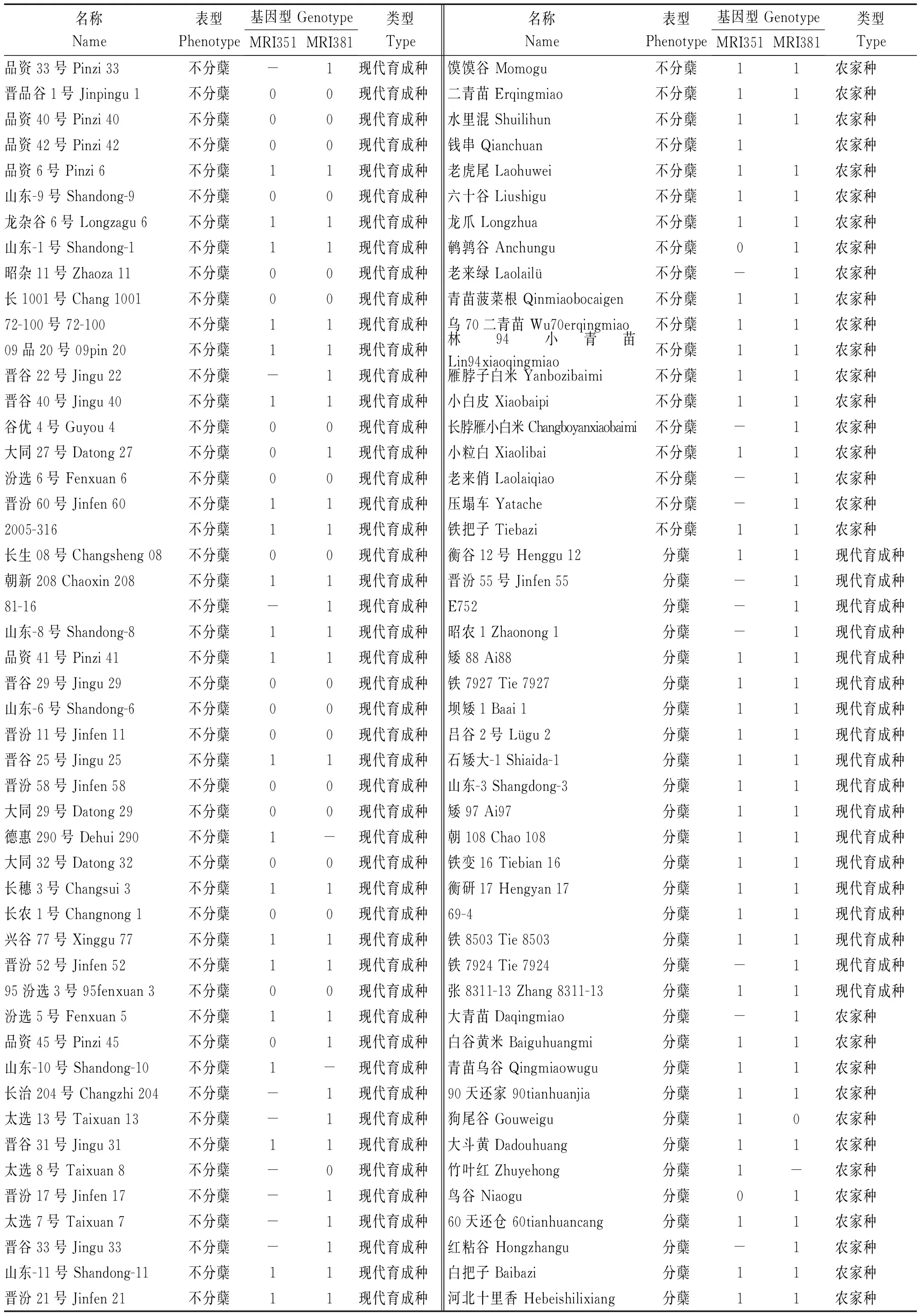
表4(续)
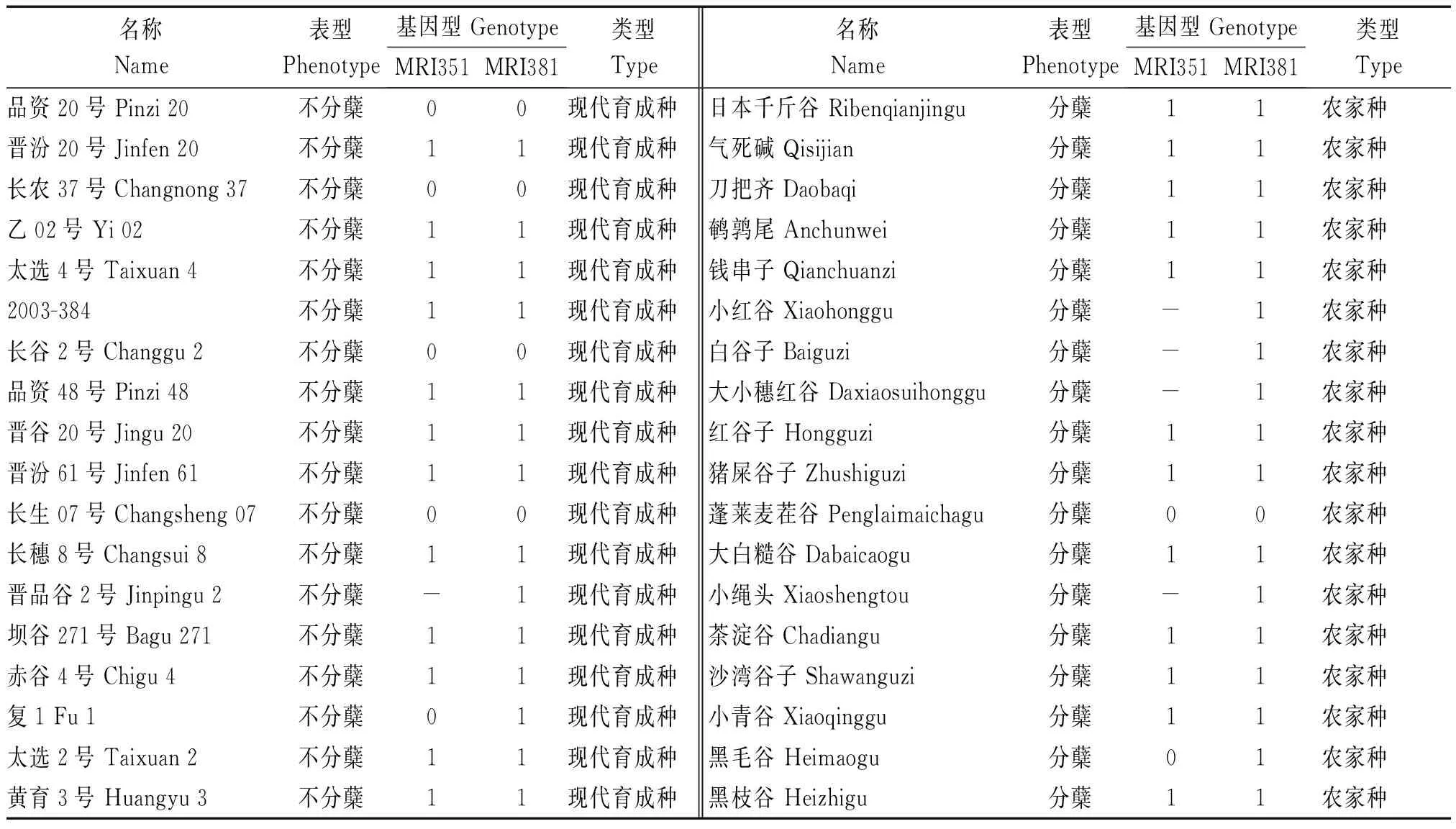
表4(续)

表5 分蘖数与标记间的相关性分析Tab.5 Correlation analysis between tiller number and markers
注:**表示差异极显著(P<0.01)。
Note:**indicate extremely significant difference (P<0.01).
3 讨论
本研究基于简化基因组测序技术(RAD-seq),以2个F2群体(Cross AJ和Cross HC)为主要研究对象,共鉴定了8个控制分蘖数的QTL。其中,qAJTN1、qAJTN5、qAJTN7-2、qAJTN9和qHCTN5等5个QTL为本研究新鉴定的位点,其余3个QTL(qAJTN7-1、qAJTN7-3和qHCTN7)与Jia等[26]和Mauro-Herrera等[3]检测的QTL相一致。
在本研究中,鉴定的较大效应位点qAJTN5(9.8%)的临近区域,Doust等[24]检测到的分蘖相关QTL可解释表型变异率为28.1%,Mauro-Herrera等[3]检测到的分蘖相关QTL(Br5c)可解释表型变异率为23.6%,Zhang等[27]检测到的分蘖相关QTL(qtn5)可解释表型变异率为25.72%和11.31%,这些QTL效应值都比较大,这很可能是由于控制同一性状的QTL成簇分布或群体差异造成的。这些结果也暗示,在谷子第5染色体存在稳定的控制分蘖相关的QTL,相关QTL区间是发掘控制分蘖相关基因的关键区间。
同时,数量性状QTL定位准确性很容易受作图群体和环境因素影响。同一性状QTL在不同群体(F2、F2∶3、RIL和NIL)中效应大小可能不同,相对于F2和F2∶3群体来说,由于RIL和NIL群体能进行多年多点重复试验,其QTL定位结果更加可靠、稳定[3, 24-25, 49]。F2群体的表型数据不能重复,因此,利用F2群体鉴定的QTL效应需要用高级群体进一步验证。尽管如此,目前在农作物中也有一些利用F2群体定位QTL的研究报道[50-52],尤其是结合下一代测序技术能对目标性状进行快速定位[53-54]。本研究采用下一代测序技术,分别利用F2大群体(543个单株)和小群体(131个单株)对控制分蘖数的QTL进行定位研究,与前人研究相比,有一致位点QTL(qAJTN7-1、qAJTN7-3和qHCTN7),也有临近位点QTL(qAJTN5),比较而言,利用大群体检测到了更多与前人研究结果相同或相近的位点,而利用小群体定位的相同或相近位点较少,这表明基于F2大群体的简化基因组测序定位QTL可靠性较F2小群体更强,至于F2大群体与高级群体间的效率差异,还需要进一步利用该F2群体的重组近交系群体进行多年多点试验来验证。
InDel标记具有带型简单、密度高、变异稳定、多态性强、易检测等特点,被广泛应用于作物种质资源品种鉴定、遗传多样性分析和分子标记辅助育种中。赵庆英等[55]利用晋谷21重测序结果开发了一个晋谷21特异的InDel标记2G5501976,该标记可快速鉴定该品种是否为晋谷21或其衍生品种。姜童等[56]利用25对InDel标记对8个鲁西南地区簇生朝天椒品种构建InDel分子遗传图谱,从而对不同品种的相似度进行评价。田希辉等[57]开发了11个与白菜苗期TuMV-C4抗性连锁的InDel标记,这些标记对高抗单株选择的准确率达78%以上,这为白菜TuMV抗性分子育种奠定了良好基础。本研究开发的InDel标记MRI351和MRI381,统计分析表明,与分蘖性状紧密连锁,这为分蘖基因的克隆和分蘖型谷子分子标记辅助育种奠定了基础。
