单甲基化表没食子儿茶素没食子酸酯的化学合成❋
2019-01-04万升标
刘 军, 孙 玥, 彭 凯, 万升标❋❋
(1. 中国海洋大学医药学院,海洋国家实验室海洋药物与生物制品功能实验室,山东 青岛 266003; 2. 安徽农业大学茶与食品科技学院,茶树生物学与资源利用国家重点实验室,安徽 合肥 230036;)
儿茶素是茶叶中最重要的生理活性物质,主要成分有四种(见图1),分别为表儿茶素(1,EC)、表没食子儿茶素(2,EGC)、表儿茶素没食子酸酯(3,ECG)、表没食子儿茶素没食子酸酯(4,EGCG)。其中,EGCG 含量最高,占儿茶素80%左右。EGCG 是儿茶素中抗氧化作用最强的一种成分,属非酶抗氧化剂,它的抗氧化活性是维生素E的20倍,具有抗肿瘤、清除体内自由基、抗炎、抗突变、降血脂、抗衰老及减少肝损伤等生物活性[1-4]。
甲基化 EGCG 是指将 EGCG 苯环上的酚羟基转化为甲基醚而形成的一系列衍生物。1982年,Saijo[5]首次从绿茶中分离出了表没食子儿茶素3-O-甲基没食子酸酯(5,3″-Me-EGCG)。1999 年,Sano等[6]于从乌龙茶中分离出了3″-Me-EGCG (5)和表没食子儿茶素 4-O-甲基没食子酸酯(6,4″-Me-EGCG)。Amarowicz等[7]从绿茶中分离出了3″-Me-EGCG和4″-Me-EGCG。
EGCG经过甲基化以后,它的脂溶性和稳定性都会得到提高。它在动物血液中的稳定性要明显高于EGCG,口服吸收率比EGCG高9倍,具有潜在的医学价值和开发应用前景[8a]。研究表明与EGCG相比,甲基化EGCG在抗过敏、降血脂等方面具有更高的生物活性。Maeda-Yamamoto等[9-11]研究表明饮用富含甲基EGCG的Benifuuki绿茶对于缓解过敏性鼻炎症状效果明显,连喝一个半月的Benifuuki绿茶可有效减轻花粉症。此外,动物实验表明3″-Me-EGCG 和4″-Me-EGCG可抑制Ⅰ型和Ⅳ型过敏性反应,且抑制效果强于EGCG[12,13]。Noritaka等[12]研究表明3″-Me-EGCG 可有效防止肝细胞在肝细胞冷藏过程中被冻伤。Kawase等[14]发现3″-Me-EGCG 在老鼠肿瘤细胞中有着很强的抗氧化性和抗细胞毒素作用,其抗氧化性比EGCG略低,但抗细胞毒素能力约为EGCG的1.5倍。Chiu等[15]研究发现3″-Me-EGCG比EGCG更能有效地抑制巨噬细胞中一氧化氮(NO)的产生及诱导型一氧化氮合成酶(I NOS)的表达,而在哮喘中,高浓度的NO可能产生各种不良反应。Kurita等[16]研究发现3″-Me-EGCG 可明显抑制血管紧张素Ⅰ转换酶的活性, 而Benifuuki茶抑制高血压的效果也远远高于其他不含甲基化EGCG的茶。
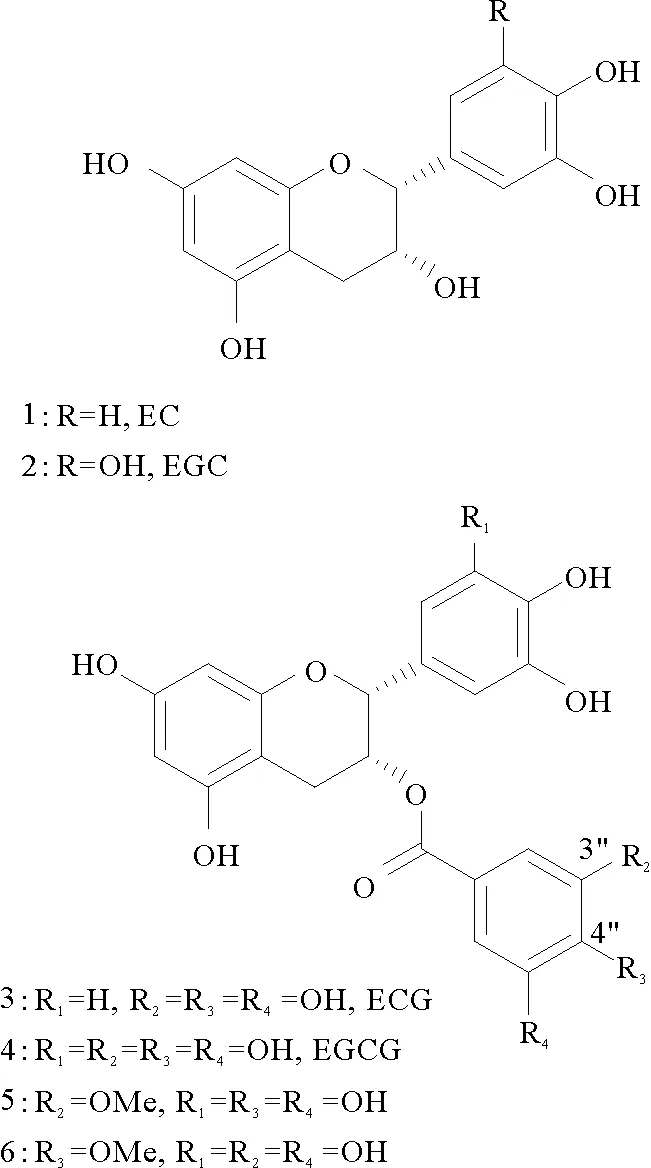
图1 EGCG及其衍生物Fig.1 EGCG and its derivatives
尽管甲基化EGCG尤其是3″-Me-EGCG 和4″-Me-EGCG 生理活性显著,稳定性和口服生物利用度与EGCG相比明显提高,潜在的药物开发价值巨大,但是茶叶中天然的甲基化EGCG 含量低,很难满足大规模生产的需要。日本的科研工作者发现在日本42个茶树种质中3″-Me-EGCG 的含量介于微量与 1.516%之间,其中含量大于 1%的茶树种质有 3 个[8b]。此外,有研究调查发现甲基化儿茶素在茶叶中的含量受到茶树品种、季节、鲜叶成熟度以及加工工艺不同而存在显著差异。总体来讲,含有甲基化 EGCG 的茶树品种较少,在含有甲基化 EGCG 的茶树品种中其含量也很低,最高不超过干重的 2%,因此直接从茶叶中分离提取来获得甲基化 EGCG面临困难。
2006年,Wan 等[17]利用苄基保护基团完成了3″-Me-EGCG 和4″-Me-EGCG的全合成工作,但是路线较长,总产率较低,不适于大规模制备。半合成方面主要分为两种策略:一是在甲基供体存在的情况下,在适当的环境中通过一步反应形成甲基化儿茶素,此路线条件控制比较困难,反应的产物较多,不易分离[4];二是利用保护基团先对酚羟基进行保护,最后脱去保护基团即可得到儿茶素的单甲基化衍生物,应用此策略的合成工作[18-19],已有报道。本文以没食子酸丙酯以及EGCG为原料,以苄基为保护基完成了3″-Me-EGCG (5)和4″-Me-EGCG(6)的化学合成,该方法比已有方法产率高,甲基化选择性高且原料经济易得。
1 实验部分
1.1 仪器与试剂
JEOL JNM-EPC 500核磁共振仪(内标TMS); JAS-COP-1020 旋光仪,Laborota旋转蒸发器。绿茶粗提物(陕西森弗高科实业有限公司);没食子酸丙酯, 98%,北京百灵威科技有限公司;溴化苄,化学纯,CP,国药集团化学试剂有限公司;4-甲氧基苯甲醛二甲缩醛,98%,北京百灵威科技有限公司;其他试剂均为市售化学纯或分析纯,除特别说明外,不经处理直接使用。
1.2 目标化合物的合成
如图2所示,先将EGCG用溴化苄在碳酸钾的条件下进行全苄基化保护,然后在碳酸钾和乙二醇二甲醚(DME)的条件下制得五苄基化(-)-EGC (2a)。
如图3所示,以没食子酸丙酯(7)为原料,用4-甲氧基苯甲醛二甲缩醛将邻羟基进行保护[20],然后用碘甲烷进行甲基化得到化合物9,用2 M的HCl溶液脱除保护基后将酚羟基用苄基保护得到化合物11,化合物11经碱水解制成化合物12,将化合物12与已经制得的苄基化(-)-EGC (2′) 酯化反应得到化合物13,最后将化合物13在氢气和氢氧化钯碳的条件下还原得到产物3″-Me-EGCG(5), 核磁数据与文献报道一致[17]。
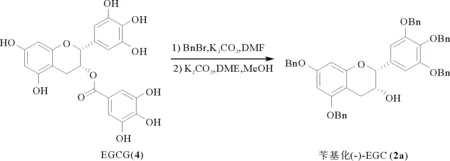
图2 苄基化(-)-EGC (2a)的制备Fig.2 The preparation of benzylated (-)-EGC 2a)

图3 3″-Me-EGCG (5)的合成Fig.3 The synthesis of 3″-Me-EGCG (5)
如图4所示,以没食子酸丙酯(7)为原料,经全乙酰化、选择性甲基化、去乙酰化得到化合物16,之后再经苄基化、水解、酯化得到化合物19,最后在氢气和氢氧化钯碳的作用下还原得到4″-Me-EGCG (6), 核磁数据与文献报道一致[17]。
1.3 目标化合物的制备
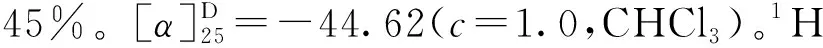
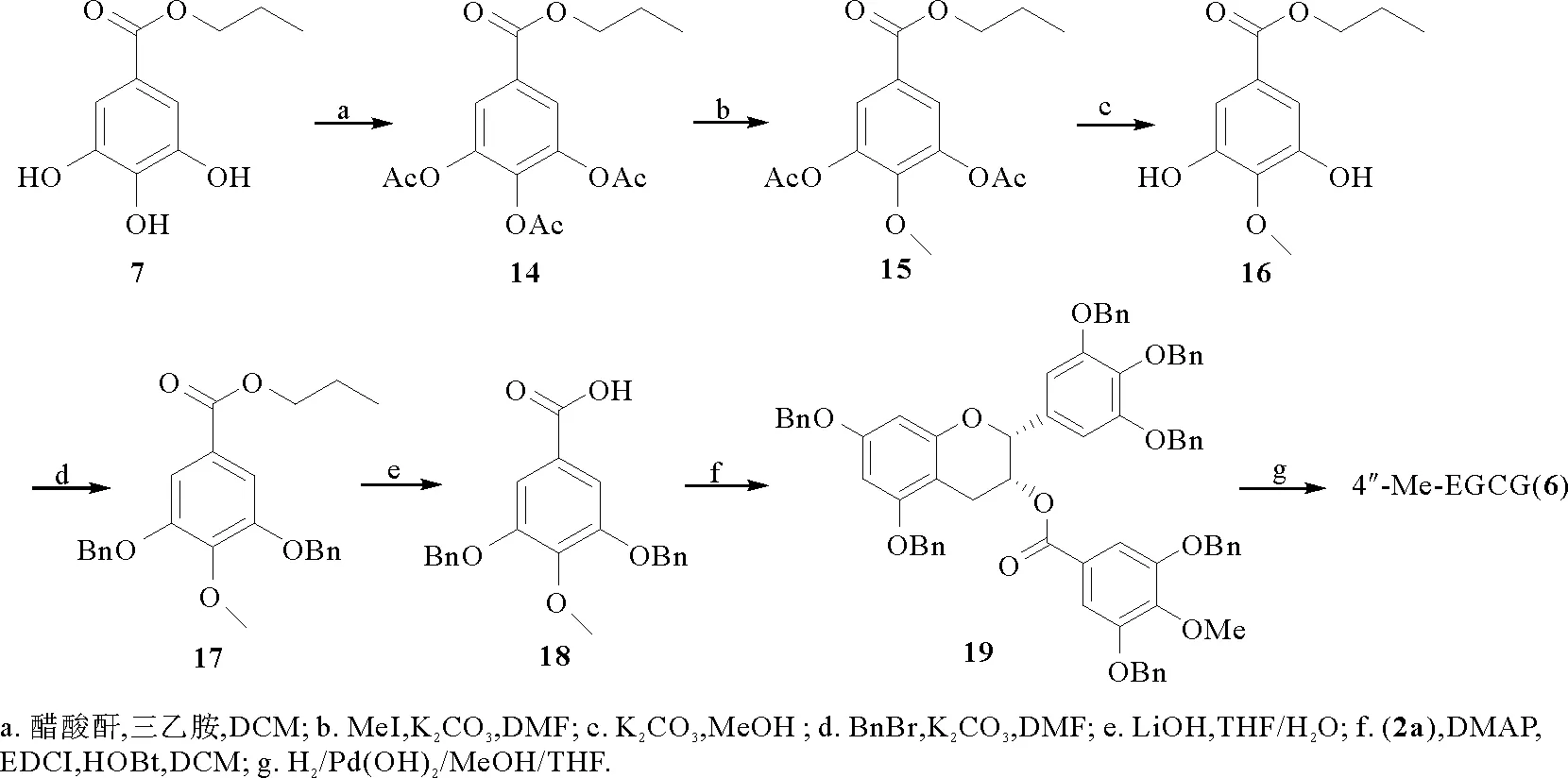
图4 4″-Me-EGCG (6)的合成Fig.4 The synthesis of 4″-Me-EGCG (6)

1.3.2 1,3-苯并二茂-5-羧酸-7-羟基-2-(4-甲氧基苯基)-,丙酯(8)的合成 向装有分水器和冷凝管的圆底烧瓶中加入没食子酸丙酯(0.9 g, 4.24 mmol),4-甲氧基苯甲醛二甲缩醛(2.30 mL, 12.72 mmol),一水合对甲苯磺酸(10 mg)和甲苯(20 mL)。氮气保护下加热回流,搅拌反应2 h, 将溶剂不断从分水器中除去,TLC监测反应完全后,冷却至室温,加入二氯甲烷,将反应液倒入分液漏斗,用40 mL饱和碳酸钠溶液洗涤,二氯甲烷萃取(20 mL×3),收集有机相,无水硫酸镁干燥,过滤,蒸干,柱层析(石油醚∶乙酸乙酯=10∶1),得白色固体1.26 g,产率90%。1H NMR (500MHz, CDCl3)δ(ppm): 1.02 (3H, t), 1.70~1.95 (2H, m), 3.81(s, 3H), 4.25 (2H, t),6.95 (d,J=8.5 Hz, 2H), 6.96 (s, 1H), 7.15 (s, 1H), 7.38 (s, 1H), 7.45 (d,J=8.5 Hz, 2H);13C NMR (126MHz, CDCl3)δ(ppm): 164.2, 161.3, 148.9, 138.9, 138.7, 128.0, 127.3, 124.1, 114.1, 111.9, 102.9, 68.4, 55.4, 21.5, 10.2。
1.3.3 1,3-苯并二茂-5-羧酸-7-甲氧基-2-(4-甲氧基苯基)-,丙酯(9)的合成 将化合物8(600 mg, 1.82 mmol)加入到10 mL DMF中,氮气保护下加入碘甲烷(0.17 mL, 2.73 mmol),碳酸氢钠(0.23 g, 2.73 mmol),室温搅拌3 h,TLC监测反应完全,将反应液倒入20 mL冰水中,过滤,将固体用二氯甲烷溶解,滤液用乙酸乙酯(10 mL×2)萃取,合并有机相,无水硫酸镁干燥,过滤,蒸干,柱层析(石油醚∶乙酸乙酯=30∶1),得白色固体592 mg, 产率94%。1H NMR (500MHz, CDCl3)δ(ppm): 1.02 (3H, t), 1.70~1.95 (2H, m), 3.81 (s, 3H), 3.83 (s, 3H), 4.25 (2H, t),6.95 (d,J=8.5 Hz, 2H), 6.96 (s, 1H), 7.15 (s, 1H), 7.38 (s, 1H), 7.45 (d,J=8.5 Hz, 2H);13C NMR (126MHz, CDCl3)δ(ppm): 164.3, 161.3, 148.9, 138.9, 138.7, 128.0, 127.3, 124.1, 114.1, 111.9, 102.9, 68.4, 55.9, 55.4, 21.5, 10.2。
1.3.4 3,4-二羟基-5-甲氧基苯甲酸丙酯 (10)的合成 将化合物9(400 mg, 1.16 mmol)溶于10 mL四氢呋喃中,逐滴加入1.67 mL HCl溶液,室温搅拌反应2 h,TLC监测反应完全,加入饱和碳酸氢钠溶液淬灭反应,反应液浓缩后用乙酸乙酯(10 mL×3)萃取,合并有机相,无水硫酸镁干燥,过滤,蒸干,柱层析(石油醚∶乙酸乙酯=10∶1),得油状产物248 mg, 产率94%。1H NMR (500 MHz, CDCl3)δ(ppm): 1.02 (3H, t), 1.70~1.95 (2H, m), 3.83 (s, 3H), 4.25 (2H, t),6.95 (s, 1H), 7.21 (s, 1H);13C NMR (126MHz, CDCl3)δ(ppm): 164.3, 145.9, 136.9, 134.7, 124.4, 111.9, 104.9, 68.4, 55.9, 21.5, 10.2。
1.3.5 3,4-二苄氧基-5-甲氧基苯甲酸丙酯(11)的合成 将化合物10(300 mg,1.33 mmol),碳酸钾(1.8 g,13.3 mmol)加入到10 mL DMF中,氮气保护下加入溴化苄(1.57 mL,13.3 mmol),室温搅拌反应过夜,TLC监测反应完全,将反应液倒入20 mL冰水混合物中,乙酸乙酯(20 mL×3)萃取,合并有机相,无水硫酸镁干燥,过滤,柱层析(石油醚∶乙酸乙酯=20∶1),得白色固体490 mg,产率:90%。1H NMR (500 MHz, CDCl3)δ(ppm): 7.43 (d,J=6.6 Hz, 4H), 7.38 (t,J=7.2 Hz, 3H), 7.34 (d,J=7.1 Hz, 1H), 7.32~7.27 (m, 4H), 5.14 (s, 2H), 5.11 (s, 2H), 4.26 (t,J=6.7 Hz, 2H), 3.89 (s, 3H), 1.79 (dd,J=14.2, 7.1 Hz, 2H), 1.02 (t,J=7.4 Hz, 3H);13C NMR (126 MHz, CDCl3)δ(ppm): 164.3, 153.4, 152.3, 141.0, 137.9, 137.3, 128.8, 128.5, 128.3, 128.0, 126.5, 108.4, 107.2, 74.5, 70.7, 68.4, 56.5, 21.5, 10.3。
本文所述的命令、配置文件等都基于RHEL6.3(Redhat Enterprise Linux 6.3),不同 Linux发行版之间或许有细微的差别,这些差异不在本文的探讨范围之内。
1.3.6 3,4-二苄氧基-5-甲氧基苯甲酸(12)的合成 将化合物11(380 mg, 0.94 mmol)用10 mL四氢呋喃∶水=5∶1的混合溶液溶解,加入氢氧化锂(90 mg, 3.76 mmol)室温下搅拌反应过夜,TLC监测反应完全,用1 mol·L-1的HCl溶液调pH至4~5,乙酸乙酯(10 mL×3)萃取,无水硫酸镁干燥,过滤,蒸干,得白色固体322 mg,产率95%。1H NMR (500 MHz, DMSO)δ(ppm): 7.45 (d,J=7.3 Hz, 2H), 7.42~7.36 (m, 4H), 7.34 (dd,J=4.2, 2.6 Hz, 2H), 7.32~7.28 (m, 3H), 7.26 (d,J=1.5 Hz, 1H), 5.16 (s, 2H), 5.01 (s, 2H), 3.83 (s, 3H)。13C NMR (126 MHz, DMSO)δ(ppm): 167.34 (s), 153.45 (s), 152.29 (s), 141.02 (s), 137.89 (s), 137.28 (s), 128.85 (s), 128.54 (s), 128.31 (d,J=3.3 Hz), 128.03 (s), 126.49 (s), 108.45 (s), 107.19 (s), 74.54 (s), 70.66 (s), 56.47 (s)。
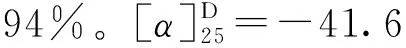
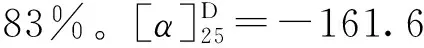
1.3.9 3,4,5-三乙酰氧基苯甲酸丙酯(14)的合成 冰浴下,将没食子酸丙酯(1 g,4.7 mmol),醋酸酐(7 mL,70.5 mmol)和三乙胺(3.9 mL,28.2 mmol)溶于20 mL二氯甲烷中,缓慢升至室温搅拌反应4 h,TLC监测反应完全。将反应液用20 mL饱和碳酸氢钠溶液水洗,分液,收集有机相,无水硫酸镁干燥,过滤,蒸干,柱层析(石油醚∶乙酸乙酯=20∶1),得到白色固体1.53 g,产率96%。1H NMR (500 MHz, CDCl3)δ7.79 (s, 2H), 4.27 (t,J=6.7 Hz, 2H), 2.30 (s, 9H), 1.77 (dd,J=14.2, 7.1 Hz, 2H), 1.00 (t,J=7.4 Hz, 3H)。13C NMR (126 MHz, CDCl3)δ(ppm): 167.6, 166.4, 164.4, 143.4, 138.5, 128.7, 122.2, 67.2, 22.0, 20.6, 20.2, 10.4。
1.3.10 3,5-二乙酰氧基-4--甲氧基苯甲酸丙酯(15)的合成 将化合物14(500 mg,1.48 mmol)溶于10 mL DMF中,加入碳酸钾(613 mg,4.44 mmol)和碘甲烷(0.11 mL,1.77 mmol),40 ℃下搅拌反应3 h,TLC监测反应完全,滤掉不溶物,加入20 mL乙酸乙酯稀释,水洗,收集有机相,无水硫酸镁干燥,过滤,蒸干,柱层析(石油醚∶乙酸乙酯=10∶1),得白色固体440 mg,产率95%。1H NMR (500 MHz, CDCl3)δ(ppm): 7.67 (s, 2H), 4.25 (t,J=6.7 Hz, 2H), 3.88 (s, 3H), 2.35 (s, 6H), 1.77 (dd,J=14.2, 7.0 Hz, 2H), 1.01 (t,J=7.4 Hz, 4H) ;13C NMR (126 MHz, CDCl3)δ(ppm): 167.8, 165.5, 147.8, 142.7, 126.5, 122.3, 67.2, 61.1, 22.0, 10.3。
1.3.11 3,5-二羟基-4-甲氧基苯甲酸丙酯(16)的合成 将化合物15(800 mg, 2.58 mmol)溶于甲醇(10 mL)和水(2 mL)的混合溶液中,加入碳酸钾(1.1 g,7.97 mmol),室温搅拌反应2 h,TLC监测反应完全,蒸干溶剂,加入乙酸乙酯稀释,用稀盐酸调pH至2,水洗,分液,收集有机相,无水硫酸镁干燥,过滤,蒸干,得到化合物16(565 mg),产率97%。1H NMR (500 MHz, CDCl3)δ(ppm): 7.01 (s, 2H), 4.25 (t,J=6.7 Hz, 2H), 3.85 (s, 3H), 1.77 (dd,J=14.2, 7.0 Hz, 2H), 1.01 (t,J=7.4 Hz, 4H);13C NMR (126 MHz, CDCl3)δ(ppm): 167.4, 151.4, 141.5, 126.5, 109.8, 67.2, 61.3, 22.0, 10.4。
1.3.12 3,5-二苄氧基-4-甲氧基苯甲酸丙酯(17)的合成 合成方法同化合物11,白色固体,产率90%。1H NMR (500 MHz, CDCl3)δ(ppm): 1.01 (3H, t), 1.77 (dd,J=14.2, 7.0 Hz, 2H), 3.89 (3H, s), 4.25 (2H, t), 5.20 (4H, s), 7.15~7.59 (12H, m);13C NMR (126 MHz, CDCl3)δ(ppm): 167.8, 151.8, 144.2, 136.6, 128.4, 128.1, 127.0, 123.8, 109.8, 71.3, 67.2, 61.3, 22.0, 10.4。
1.3.13 3,5-二苄氧基-4-甲氧基苯甲酸(18)的合成 合成方法同12,白色固体,产率91%。1H NMR (500 MHz, CDCl3)δ(ppm): 3.89 (3H, s), 5.21 (4H, s), 7.28~7.52 (12H, m);13C NMR (126 MHz, CDCl3)δ(ppm): 171.3, 152.2, 144.4, 136.6, 128.5, 128.1, 127.4, 123.9, 109.8, 71.3, 61.2。


2 结果与讨论
本文以没食子酸丙酯和EGCG等为原料,以苄基为保护基,分别以53%和57%的总产率完成了3″-Me-EGCG 和4″-Me-EGCG 的半合成,所得化合物核磁数据与文献报道一致[17]。Aihara 等[18]以邻硝基苯磺酰基和烯丙基为保护基完成了3″-Me-EGCG的半合成,最终总产率为52%,本文采用的方法与其相比,在总产率上提高了1%。在4″-Me-EGCG的半合成方法中,Aihara等[18]以没食子酸烯丙酯为原料在碳酸锂条件下直接进行单甲基化,产率为68%;Lai等[20]以没食子酸丙酯为原料参照Aihara的方法得到化合物16,产率为83.0%。而Zhu等[21]以没食子酸甲酯为原料,经过全乙酰化,选择性甲基化和水解得到4位甲基化的没食子酸甲酯,三步反应总产率高达96%。本文最初尝试采用Aihara 和Lai的方法制备化合物16,但是在实际操作过程中发现酚羟基甲基化的选择性较差,难以选择性得到化合物16。因此,本文参照Zhu等人的方法进行化合物16的制备,产率达到88.4%,最后以57%的总产率得到4″-Me-EGCG(6), 与Aihara等相比提高了19%。Lai 同样采用苄基为保护基结合Aihara 的方法完成了4″-Me-EGCG 的合成工作,但是在甲基化的选择性和总产率上与本文的合成方法相比有一定的差距。
综上所述,在3″-Me-EGCG 和4″-Me-EGCG 的化学合成中,本文探索了一种高产率,原料经济易得,操作简易的新反应路线,在3″-Me-EGCG 和4″-Me-EGCG 的大规模制备上可以作为最佳选择。
3 结语
近年来,随着甲基化EGCG尤其是3″-Me-EGCG 和4″-Me-EGCG 研究的深入,其显著的生理活性和潜在的药物开发价值引起人们越来越多的关注。但是由于甲基化EGCG 在茶叶中含量低,全合成路线长,产率低,均难以大量制备。本文以没食子酸丙酯和EGCG等为原料,分别以53%和57%的总产率完成了3″-Me-EGCG 和4″-Me-EGCG 的半合成,是目前为止总产率最高的合成方法。
