用全基因组测序评价VNTR分型在泛耐药结核分枝杆菌传播中的应用
2018-12-26陈昕昶陈嘉臻张文宏
陈昕昶,陈嘉臻,张文宏
复旦大学附属华山医院感染科,上海 200040
结核病是由结核分枝杆菌引起的传染性疾病。相比于敏感株,结核分枝杆菌耐药株的传播力是研究热点之一[1]。本课题组前期收集了55株2003—2009年于重庆市肺科医院诊断的泛耐药结核分枝杆菌(extensively drug-resistantMycobacteriumtuberculosis,XDR-TB)菌株,但由于技术上的限制,2014年仅用传统的多位点数目可变串联重复序列(variable-number tandem-repeat,VNTR)分型对其传播及成簇特征进行了分析,发现该地区XDR-TB的传播率较高[2]。
VNTR分型方法因简便易行、结果可读性强、实验室间可比性高,广泛应用于结核分枝杆菌的传播研究[3-4]。近年来,随着技术的进步、测序成本的降低,全基因组测序(whole-genome sequencing,WGS)技术开始用于研究结核分枝杆菌的传播,已有多项研究证明WGS较VNTR分型具有更高的分辨率[5-6]。但目前应用WGS鉴定结核病传播的研究主要集中于敏感菌株及欧美菌株[7-9],我国研究较少[10],且尚未在XDR-TB中进行相关研究。因此,本研究应用WGS对上述菌株进行重新分析,并评价VNTR分型判断XDR基因型分布及成簇特征的准确性。
1 材料与方法
1.1 研究对象
收集2003—2009年重庆市肺科医院诊断的55株XDR-TB菌株。该医院于2003—2009年共诊断 2 727 例结核病,其中624例为多重耐药结核分枝杆菌(multidrug-resistantMycobacteriumtuberculosis,MDR-TB),85例为XDR-TB(已去重)。2015年共回溯、成功复苏、完成药敏试验和VNTR分型的菌株有95株[2]。本次成功复苏并完成WGS的XDR-TB菌株有55株。
1.2 研究方法
1.2.1菌株选取对55株XDR-TB菌株进行复苏,并进行DNA抽提。
1.2.2VNTR分型对成功复苏并完成WGS的55株菌株进行9+3个位点的VNTR分型,纳入的9个位点为QUB-11b、QUB-18、QUB-26、MIRU26、Mtub21、Mtub04、MIRU31、MIRU40、VNTR2372,3个高变位点为VNTR3232、VNTR3820、VNTR4120[11]。聚合酶链反应(polymerase chain reaction,PCR)产物用2%琼脂糖凝胶分析。
计算每个位点重复序列的拷贝数,在VNTRplus在线分析平台(http://www.VNTRplus.org/MIRU/index.faces)上,用UPGMA算法构建系统发生树。具有完全相同MIRU-VNTR基因型的结核分枝杆菌分离株≥2株即被认为成簇。
1.2.3WGS分型对所有菌株DNA进行文库构建,基于Illumina X10平台进行测序,深度至少为200×,用Bowtie 2软件将测序reads比对到H37Rv(NC_000962.3)参考序列,覆盖率为 99.21%~99.89%。用SAMtools进行SNP检测,若某位点的突变型比例在80%以上即认为是固定突变。分析菌株间的遗传距离时,去除PE、PPE等高度重复区域,GC富集序列及耐药相关基因SNP,且SNP需测得至少10个reads无正负链偏差。基于SNP结果,用MEGA 7软件以最大似然法构建系统发育树,以H37Rv为根。根据现有研究,规定遗传距离相差不超过12个SNP为成簇,即有近期传播关系[7,10]。
2 结果
2.1 菌株信息
本研究最终纳入55株2003—2009年重庆市肺科医院诊断为XDR-TB的菌株。当时患者基本资料如下:平均年龄15~82(47±18)岁,其中男性21例(38.2%)。均有4个一线抗结核药物[异烟肼(H)、利福平(R)、链霉素(S)、乙胺丁醇(E)]和3个二线抗结核药物[氧氟沙星(O)、卡那霉素(K)、卷曲霉素(C)] 的表型药敏试验结果,耐药分布特征为HRSOK 4例、HRSOC 2例、HRSOKC 5例、HRSEOK 10例、HRSEOC 6例、HRSEOKC 28例。
2.2 VNTR分型结果
55株结核分枝杆菌通过VNTR分型方法可分为45个基因型。39株为单一基因型,16株(29.1%)分别归入6个基因簇,即C1、C2、C3、C4-1、C4-2、C4-3,每个簇分别含有5、2、3、2、2、2株分离株。用UPGMA算法构建系统发育树(图1)。
2.3 WGS分型结果
根据SNP,将55株菌株分为2个谱系[12],其中53株为Lineage 2(East-Asian),2株(Q546、Q388)为Lineage 4(Euro-American)。
根据现有研究,规定遗传距离相差不超过12个SNP为成簇,可将55株菌株中的20株(36.4%)分为5个簇,即W1、W2、W4、W5、W6,分别含有6、4、5、3、2株菌株,并用MEGA 7软件以最大似然法构建系统发育树(图2)。
簇内菌株间SNP差异个数为4~21个,平均相差(12.89±3.62)个SNP。5个簇簇内SNP差异个数分别为7~21(平均13.9±3.6)、9~19(平均 11.3±3.9)、4~21(平均13.2±4.7)、12、7个。进一步比较簇内关系最接近的菌株,其两两之间相差的SNP个数为4~12个,平均(9.8±2.4)个。5个簇簇内菌株与簇外其他菌株的SNP差异个数分别为 (341.9±120.1)、(288.9±120.3)、(290.0±138.5)、(285.2±123.3)、(351.0±82.5)个(表1)。
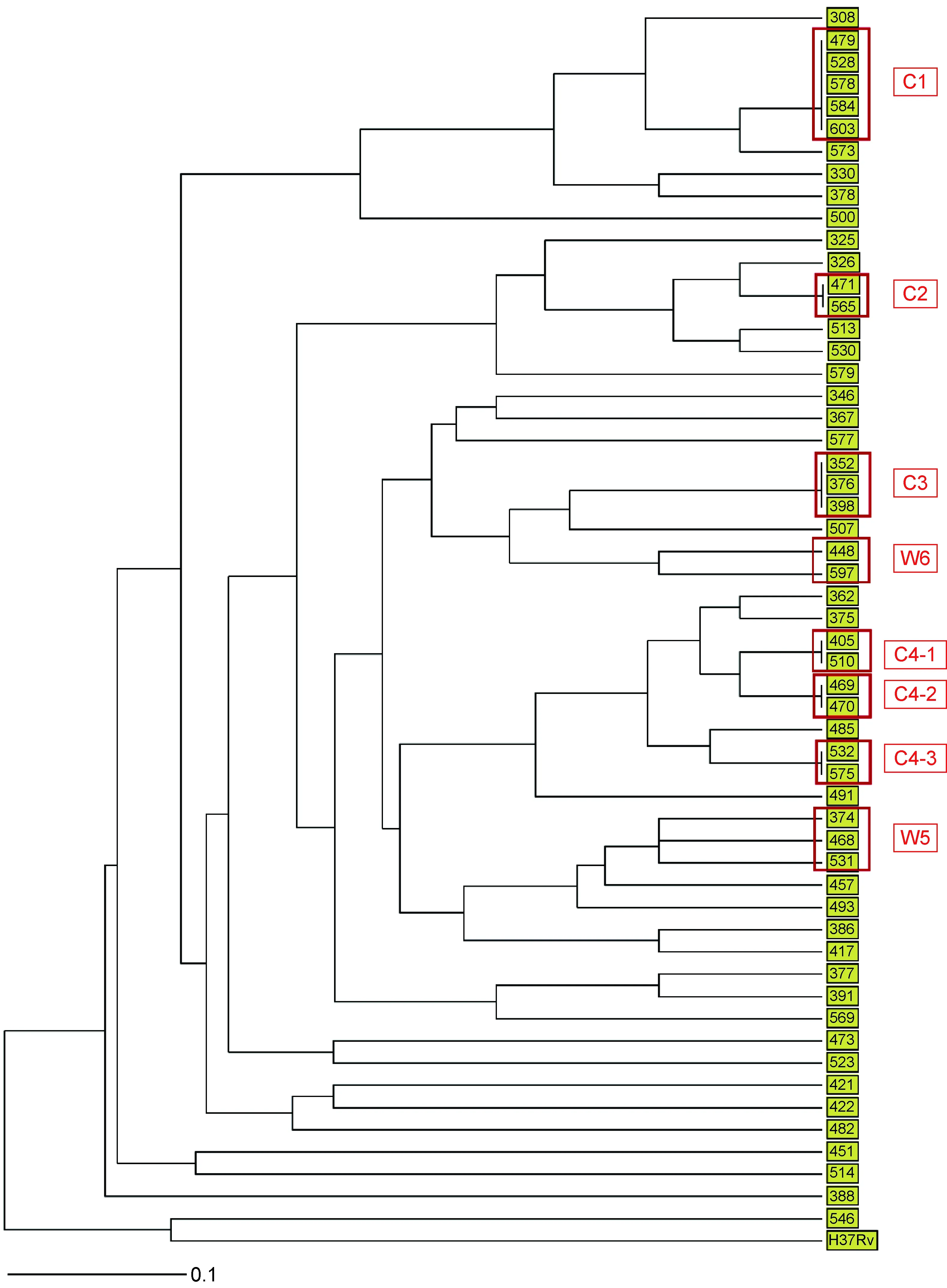
注:红色框内是用VNTR、WGS分型方法鉴定的簇,其中C簇是VNTR分型鉴定出的簇,W簇是WGS分型鉴定出的簇。
图1基于VNTR分型方法构建进化树
Fig.1PhylogenetictreebasedonVNTRtyping
2.4 VNTR分型与WGS分型结果比较
VNTR将55株菌分成6个簇,WGS分成5个簇,其中C1与W1、C2与W2、C4-1~C4-3与W4有对应关系。
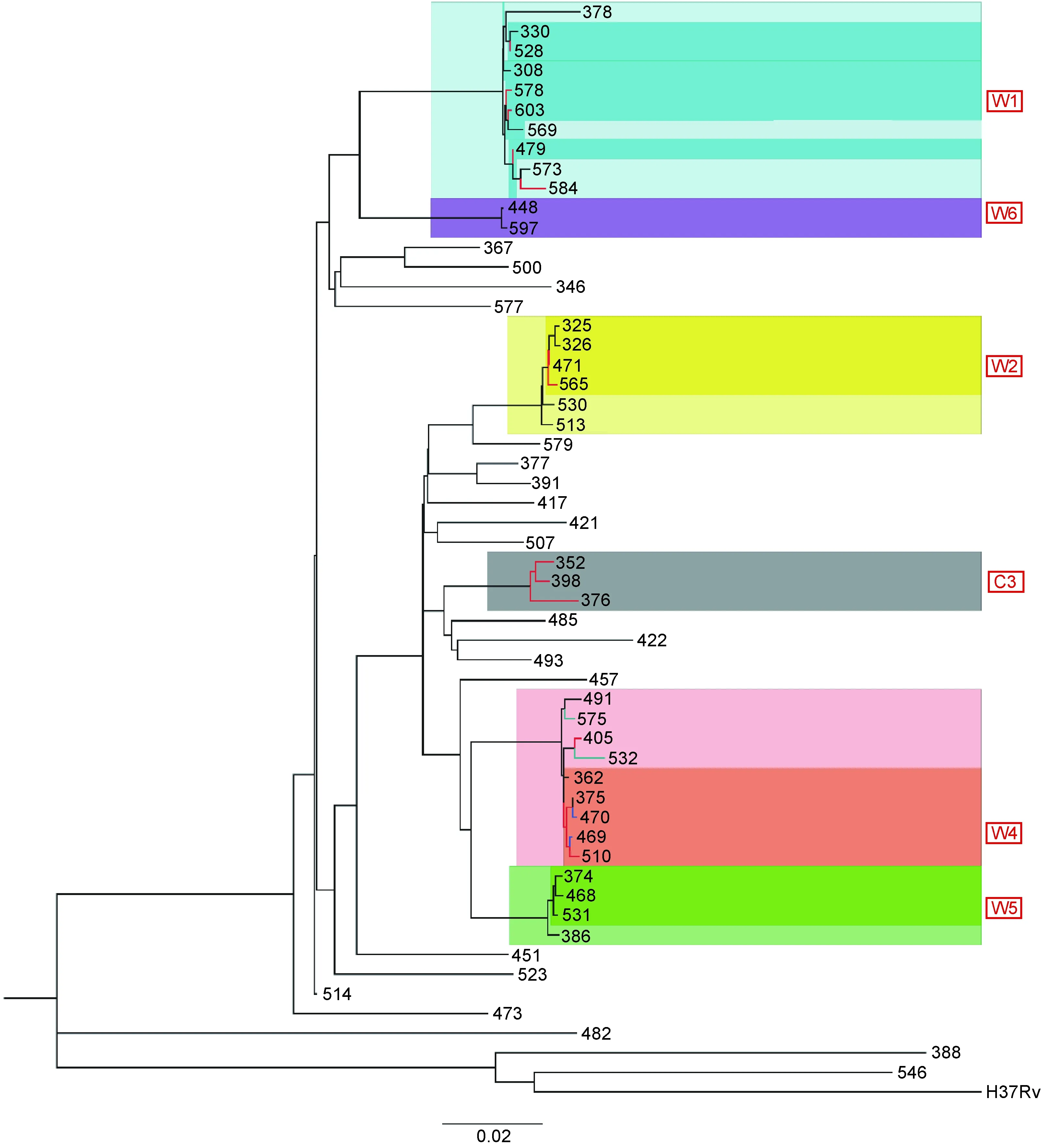
注:红色框内是用VNTR、WGS分型方法鉴定的簇,其中C簇是VNTR分型鉴定出的簇,W簇是WGS分型鉴定出的簇。
图2基于WGS分型方法构建进化树
Fig.2PhylogenetictreebasedonWGS
表1全基因组测序方法鉴定成簇的菌株及其SNP信息
Tab.1Clustersofstrainsidentifiedbywhole-genomesequencing
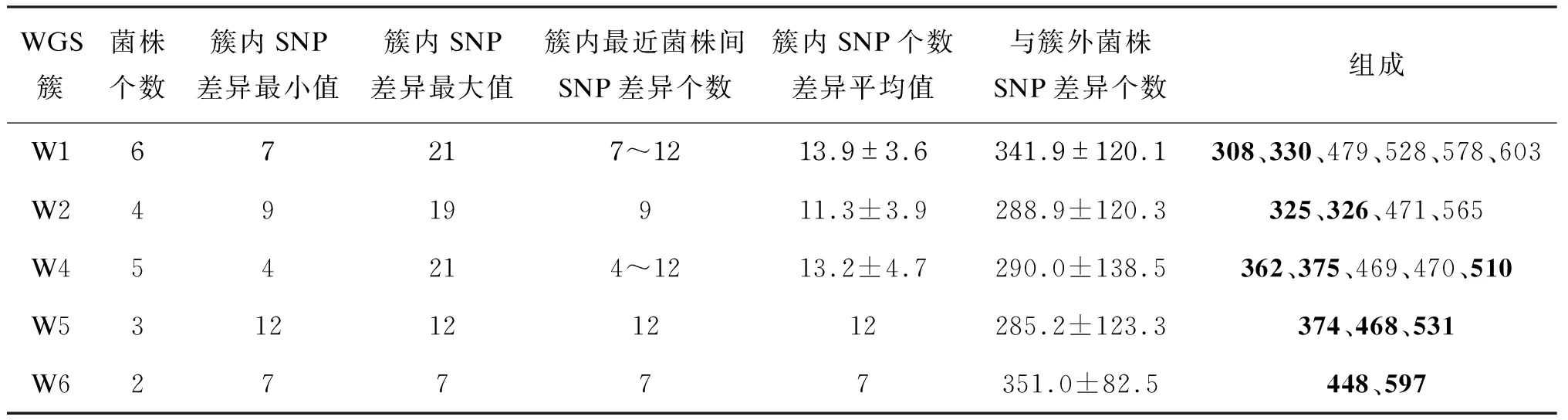
WGS簇菌株个数簇内SNP差异最小值簇内SNP差异最大值簇内最近菌株间SNP差异个数簇内SNP个数差异平均值与簇外菌株SNP差异个数组成W167217~1213.9±3.6341.9±120.1308、330、479、528、578、603W24919911.3±3.9288.9±120.3325、326、471、565W454214~1213.2±4.7290.0±138.5362、375、469、470、510W5312121212285.2±123.3374、468、531W627777351.0±82.5448、597
注:加粗的编号是WGS特有的菌株,未加粗的编号是两种分型方法共有的菌株。
VNTR分型鉴定C1中的5株为一簇,WGS分型鉴定W1中的6株为一簇,两种方法均将Q479、Q528、Q578、Q603这4株分为同一簇。但Q584仅被VNTR鉴定为C1簇,WGS提示其不成簇,且与W1中其他菌株至少相差30个SNP;Q308、Q330仅被WGS鉴定为W1,VNTR提示其不成簇,且与C1中其他菌株分别相差2和3个VNTR位点。
VNTR分型鉴定C2中的2株为一簇, WGS分型鉴定W2中的4株为一簇。W2包含了VNTR分型鉴定的C2中的Q471、Q565两株,并在此基础上多鉴定出Q325、Q326与其成簇,Q325、Q326与C2簇分别相差1和3个VNTR位点。
VNTR分型鉴定Q405、Q510为一簇,即C4-1;鉴定Q469、Q470为一簇,即C4-2;鉴定Q532、Q575为一簇,即C4-3。其中C4-1与C4-2相差1个VNTR位点,C4-3与其他两个簇相差2个VNTR位点。WGS分型鉴定W4中的5株为一簇,W4中包含了C4-2的全部菌株(Q469、Q470)和C4-1中的Q510。此外,WGS还多鉴定出Q362、Q375与其成簇,这两株与其他C4菌株相差1个VNTR位点。VNTR鉴定为一簇,但WGS提示不成簇的菌株中,C4-3中的Q532与Q575相差45个SNP;C4-1中的Q405与Q510相差29个SNP。
VNTR分型鉴定C3中的3株为一簇(Q352、Q376、Q398),WGS提示其两两之间相差29、59、64个SNP,因此不成簇。此外,WGS新鉴定了2个簇,W5包含3株(Q374、Q468、Q531),其两两之间相差12个SNP、2~3个VNTR位点;W6包含2株(Q448、Q597), 相差7个SNP、 2个VNTR位点(表1~2)。
表2VNTR分型方法鉴定成簇的菌株及其SNP信息
Tab.2ClustersofstrainsidentifiedbyVNTRtyping
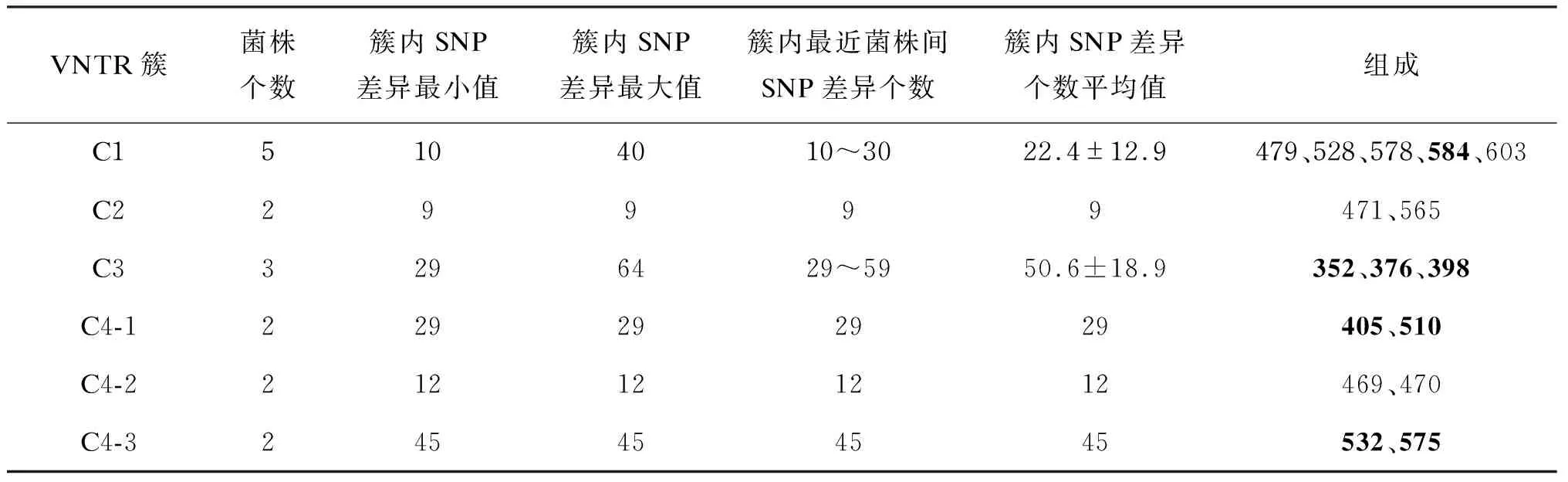
VNTR簇菌株个数簇内SNP差异最小值簇内SNP差异最大值簇内最近菌株间SNP差异个数簇内SNP差异个数平均值组成C15104010~3022.4±12.9479、528、578、584、603C229999471、565C33296429~5950.6±18.9352、376、398C4-1229292929405、510C4-2212121212469、470C4-3245454545532、575
注:加粗的编号是VNTR分型特有的菌株,未加粗的编号是两种分型方法共有的菌株。
共有8株同时被两种方法鉴定为成簇,8株仅VNTR鉴定其成簇,12株仅WGS鉴定其成簇,其余27株两种方法均提示不成簇。VNTR与WGS分型的一致性为 63.6%(35/55)。与WGS相比,VNTR分型的特异度为 77.1%,灵敏度为 40.0%,阳性预测值为 50.0%,阴性预测值为 69.2%(表3)。
表3WGS与VNTR分型鉴定成簇的一致性比较
Tab.3ConsistencycomparisonbetweenWGSandVNTR
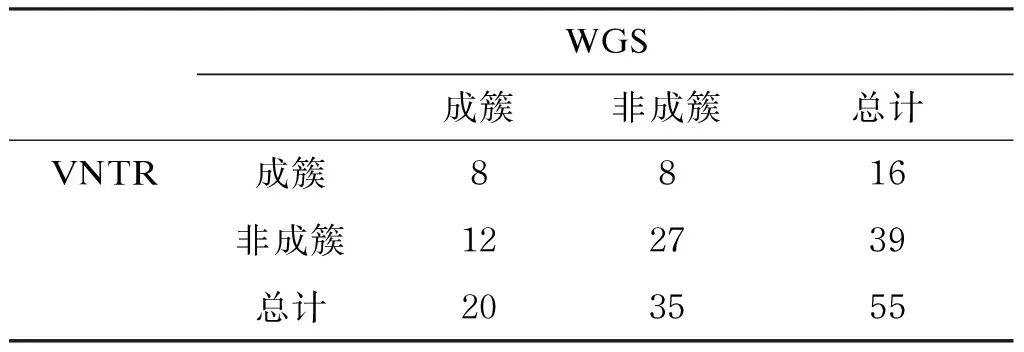
WGS成簇非成簇总计VNTR成簇8816非成簇122739总计203555
3 讨论
VNTR分型与WGS分型相比,有20株成簇判断结果不一致,其中8株仅VNTR鉴定其成簇,12株仅WGS鉴定其成簇。8株仅VNTR鉴定成簇的菌株中,错误地将C3中的3个菌株分为一簇(Q352、Q376、Q398),WGS提示Q398、Q352分别与Q376相差59、64个SNP。这一现象已被多项研究报道,即VNTR分型一致的簇内SNP差异很大[6,13],因此公认在鉴定传播时WGS比VNTR分辨率高。
12株仅WGS鉴定成簇的菌株中,W1中的Q308和Q330、W2中的Q326与C1簇、C2簇的其他菌株相差1或2个VNTR位点。WGS新鉴定了2个簇,共5个菌株(Q374、Q468、Q531、Q448、Q597),其簇内菌株间相差2个VNTR位点。这可能与VNTR多变位点的进化有关,同时也提示不能因为菌株间仅相差1~2个VNTR位点就排除近期传播[14]。现有研究多是在VNTR分型一致的菌株中进行WGS分型,很少在VNTR不一致的菌株中直接使用WGS分型并评估其效能,因此没有全面估计VNTR分型的灵敏度。
以往研究通过将WGS数据与流行病学调查结果结合,发现有流行病学联系的MDR菌株之间最多相差12个SNP,因此定义菌株间差异小于12个SNP为近期感染[7,10,15]。但是,本研究中纳入的均为XDR菌株,超过一半的菌株同时对异烟肼、利福平、乙胺丁醇、链霉素、二线注射类药物、氟喹诺酮类药物、丙硫异烟胺、对氨基水杨酸8类药物耐药,且对乙硫异烟胺和对氨基水杨酸这两种药物来说,目前已知的耐药基因仅能解释不到60%的表型耐药[16-18],其具体耐药机制及是否有补偿突变并不明确,导致在药物压力下筛选出的耐药相关基因及其补偿突变无法被过滤,可能因此造成菌株间SNP差异个数较多,即遗传距离较远。采用一个恒定的差异SNP阈值是适用于所有情况,还是仅限于某些特定情境,还有待进一步研究[14]。本研究尝试将55株菌株进行两两比对,构建SNP差异个数的矩阵,并绘制散点图(图3);但是由于缺乏流行病学数据,无法通过此矩阵定义判断近期传播的SNP阈值。
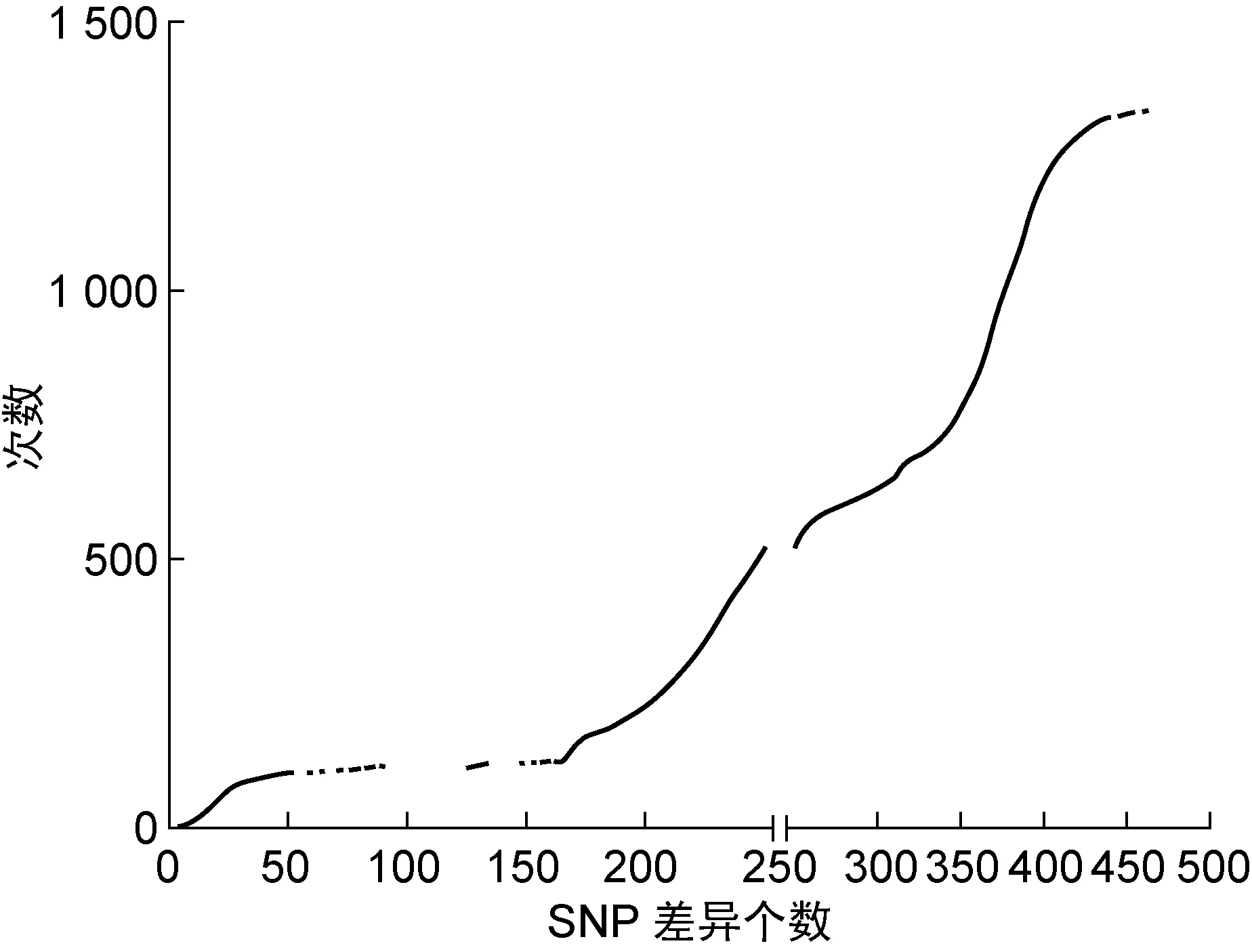
图3所有菌株间两两比较SNP差异个数
Fig.3PairwiseSNPdifferencesamong55strains
由于结核分枝杆菌的遗传学特性,其可随机发生自发突变,且回复突变极少见。假设A同时将其菌株传给B、C两人,A菌在B、C两人体内分别进化,根据现有研究,则B与A、C与A的遗传距离小于12个SNP;但若直接比较B、C两菌,其差距最大可能达24个SNP。由于本研究中纳入的是单中心取样的菌株,并非疾病预防控制中心所获得的某地区的全部菌株,很有可能存在上述A病例丢失的可能性,造成具有近期传播关系的菌株间的遗传距离较大。因此,本研究中以菌株间差异小于12个SNP为近期感染这一标准,可能过于严苛。若定义菌株间差异小于24个SNP为近期感染,则可将55株菌株中的28株(50.91%)分入5 簇。放宽标准后,与W1很接近的菌株Q569、Q573,分别与W1中的菌株最小相差16、23个SNP;与W2很接近的菌株Q513、Q530,分别与W2中的菌株最小相差17个SNP;与W4很接近的菌株Q491、Q575、Q405,分别与W4中的菌株最小相差23、15、20个SNP,且菌株Q405与C4-1中的Q510有一致的VNTR分型。
本研究存在以下不足。2003—2009年诊断为XDR的患者痰标本培养物于 -80 ℃ 冰箱中保存至今,时间久远,仅有55株复苏成功。本研究纳入的是单中心收集的菌株,并非疾病预防控制中心所获得的某地区的全部菌株,且没有流行病学调查结果,因此缺失较多病例,即缺失传播的中间环节,无法构建传播链。成簇菌株采样时间跨度较大,最长时间间隔为42个月,可能造成簇内菌株遗传距离增加,这一现象在以往大型研究中也有提及[8]。
本研究采用基于SNP的分型方法评价VNTR分型在XDR-TB传播研究中的应用,发现VNTR分型具有较高的特异度,但仅用此方法可能会错误估计XDR的传播性。与VNTR分型相比,WGS可更全面地鉴定出传播簇。现有研究认为MDR-TB中菌株间相差不超过12个SNP即有近期传播关系,这一临界值是否适用于XDR-TB有待进一步研究。
