甘氨酸与6-羟基嘌呤氢键作用的理论研究
2018-12-25裴玲,郝玮
裴 玲,郝 玮
甘氨酸与6-羟基嘌呤氢键作用的理论研究
裴 玲,郝 玮
(滨州学院 化工与安全学院,山东 滨州 256603)
采用密度泛函理论方法,在M062X/6-311+G(d,p)水平上对甘氨酸、6-羟基嘌呤及其复合物进行结构优化和频率计算,得到了10种稳定的复合物。分析发现,所有复合物进行基组重叠误差(BSSE)校正后的相互作用能均在-14.60~-70.56 kJ·mol-1,属于氢键能量范围;相互作用能主要由氢键贡献,其大小与氢键的数目有关。通过自然键轨道(NBO)理论分析验证了复合物中氢键的存在。分子中的原子(AIM)理论分析表明,Y···H存在键鞍点,且电子密度和Laplacian值都在氢键范围之内,所得复合物的相互作用均为闭壳层体系之间的相互作用。
密度泛函理论;氢键;NBO;AIM
1 引言
绝大多数的物理和化学现象与分子间相互作用密切相关。近年来,人们研究了多种生物小分子之间的相互作用,并揭示了其作用本质[1-9]。甘氨酸是构成蛋白质的基本单位,6-羟基嘌呤是一种用途十分广泛的碱基化合物,存在于许多动植物体中,可用于生物发酵、化学合成核苷类抗病毒药品。密度泛函理论(DFT)作为研究多电子体系电子结构的量子力学方法,目前已得到广泛应用,如分子间作用研究[5-10]、物质抗氧化活性研究[11-14]等。运用DFT研究甘氨酸与6-羟基嘌呤分子间相互作用的本质,可以从分子水平上深入了解甘氨酸与6-羟基嘌呤分子间相互作用机制,为之后新型药品的研究、设计、合成提供有价值的理论数据和信息。
2 计算方法
运用DFT的M062X方法,在6-311+G(d,p)基组水平上对甘氨酸与6-羟基嘌呤单体及其复合物进行几何结构优化,得到了10种稳定的复合物。在相同基组水平上,应用自然键轨道(NBO)理论[15]和分子内原子(AIM)理论[16-17]对甘氨酸与6-羟基嘌呤的相互作用特征和本质进行分析。计算复合物相互作用能时,考虑了基组重叠误差(BSSE),采用完全均衡校正(CP)方法在6-311+G(d,p)基组水平上完成了BSSE校正[18]。所有单体与复合物的优化计算采用Gaussin09程序[19]完成,AIM分析采用Multiwfn3.4.1程序[20]完成。
3 结果与讨论
3.1 甘氨酸-6-羟基嘌呤复合物的几何构型

图1 甘氨酸-6-羟基嘌呤复合物优化后的几何构型

图2 甘氨酸与6-羟基嘌呤复合物相互作用能ΔE图
优化得到的甘氨酸-6-羟基嘌呤复合物构型及结构参数如图1所示,BSSE校正[18]后的能量数值如图2所示。结果表明,甘氨酸-6-羟基嘌呤复合物为了达到最稳定状态,会尽可能多地形成氢键这种几何优势来降低复合物体系的能量,其中,甘氨酸和6-羟基嘌呤分子既是氢键供体又是氢键的受体。
由图1可以看出这10种复合物所形成的氢键类型及数目不尽相同,形成的氢键类型包括:O-H···O、O-H···N、O···H-N和N···H-N,氢键键长在1.688-2.209 Å,键角在130.19 º-178.37 º。复合物1、2、3、5、6、7含有一个氢键,复合物4、8、9、10含有两个氢键。
当分子间复合物只有一个氢键形成时,相互作用能的大小可以反映出氢键的强弱以及复合物的稳定性[21]。但是,当复合物中存在2个或更多氢键时,对复合物稳定性及氢键强弱的判断将更加复杂。分子中的氢键数目越多,复合物越稳定,经过BSSE校正后,发现含有二氢键复合物的相互作用能大(复合物1除外),单氢键复合物的相互作用能小,这表明复合物中形成的氢键数目与相互作用能的大小基本成正比。复合物8的相互作用能最大,是所有复合物中最稳定的。由图1可知,最稳定的复合物8中存在N-H···O和O-H···N这2种类型的氢键,其键长分别为1.867 Å、1.723 Å,形成的氢键使甘氨酸与6-羟基嘌呤分子稳定在处于近平面的位置,分析原因可能是由于甘氨酸与6-羟基嘌呤形成2个方向相反的氢键,化学键之间的斥力较小,结构处于近平面位置,比其它复合物更具有几何优势,相互作用能也就越大。综合以上,可以看出,氢键作用对甘氨酸-6-羟基嘌呤复合物稳定性起着重要作用。
将所优化的复合物前后相应键的键长、振动频率及变化数值整理列于表1。可以看出,当复合物形成氢键后,相应的O-H、N-H键键长均表现出伸长,变化范围在0.004-0.059 Å,振动频率明显减小,出现了正常的红移。分析原因主要是X-H···Y氢键中,电子由电子供体内孤对电子轨道进入电子受体内X-H的反键轨道,从而引起X-H键的伸长并导致红移氢键的出现[22]。另外,从表1可以看出,N-H···O和O-H···N型氢键键长和频率变化比N-H···N型氢键要大。这可能是因为N和O原子上的孤对电子与相邻的O-H、N-H反键轨道相互作用强,红移增大的程度与复合物相互作用能的变化趋势基本一致。
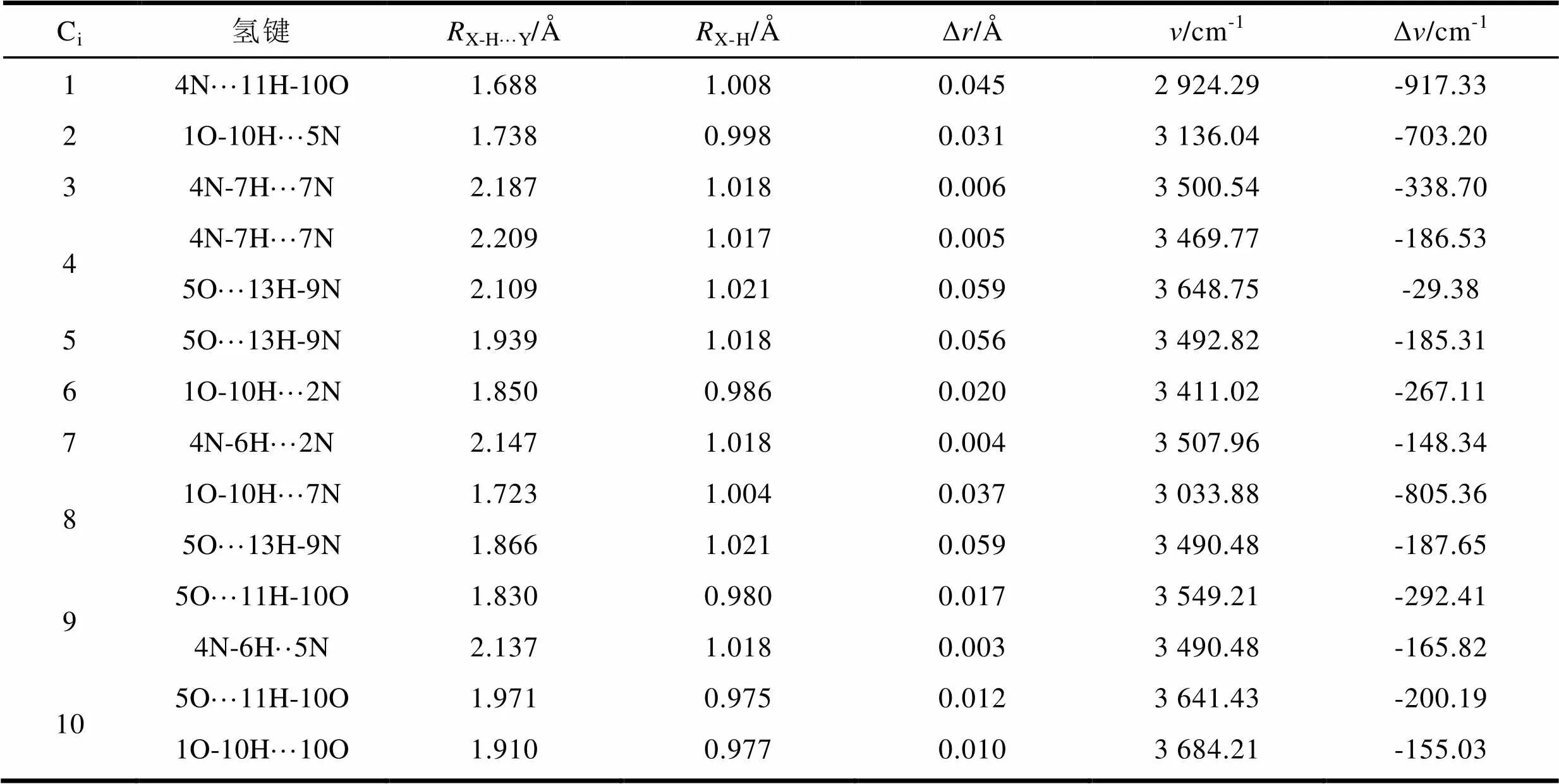
表1 M602X/6-311+G(d,p)基组水平计算的复合物氢键键长、振动频率及变化值
3.2 自然键轨道(NBO)分析
通过NBO分析所得的参数数据,可以从中找出各种分子轨道的类型、分子中原子集居数、构成分子内、分子键相互作用。运用NBO分析分子内轨道间相互作用,为氢键的存在提供理论依据。表2给出了甘氨酸-6-羟基嘌呤复合物各构型中电子供体(donor)轨道、电子受体(acceptor)轨道及其相互作用的二阶稳定化能(2)。NBO理论表明,电子供体轨道与电子受体轨道间相互作用(2)大于零,所得复合物分子之间存在氢键,因此,可以判定10种复合物中均存在氢键。
3.3 分子内原子(AIM)理论分析
根据Bader提出的“分子内原子”理论[16,17],分子电子密度分布的拓扑性质取决于电子密度的梯度矢量场▽pb和Laplacian量▽2pb,电荷密度的Laplacian量▽2()是▽()的一阶导数,▽2()=1+2+3,i为临界点处电荷密度的矩阵本征值。若矩阵的3个本征值中1个为正值、2个为负值,可将其记为(3,-1)关键点,称作键鞍点(BCP)。键鞍点处的()可用来描述键的强度,一般来说,()越大,该处化学键的强度越强。在X-H···Y体系中,如果其电子密度和Lpalcaina值分别在0.002 0-0.040 0 a.u.和0.024 0-0.139 0 a.u.范围内,一般就认为H···Y间存在键鞍点。我们对10种甘氨酸-6-羟基嘌呤复合物进行了电子密度拓扑分析,结果如表3所示。
从表3中可见,▽2BCP、BCP+BCP的值都是正的,由此可判定10种复合物均存在氢键弱相互作用。另外,大多数BCP也大于零,因此这些复合物相互作用为闭壳层体系之间的相互作用。
从表3中还可看出,双氢键复合物4、8、9、10中临界点处的平均电荷密度分别为0.017 2 a.u.和0.038 8 a.u.、0.024 4 a.u.、0.024 2 a.u.,Lpalcaina值均处于0.060 3- 0.116 6 a.u.范围内,所以这些复合物均存在键鞍点(BCP)。

表2 甘氨酸与6-羟基嘌呤复合物的自然键轨道分析
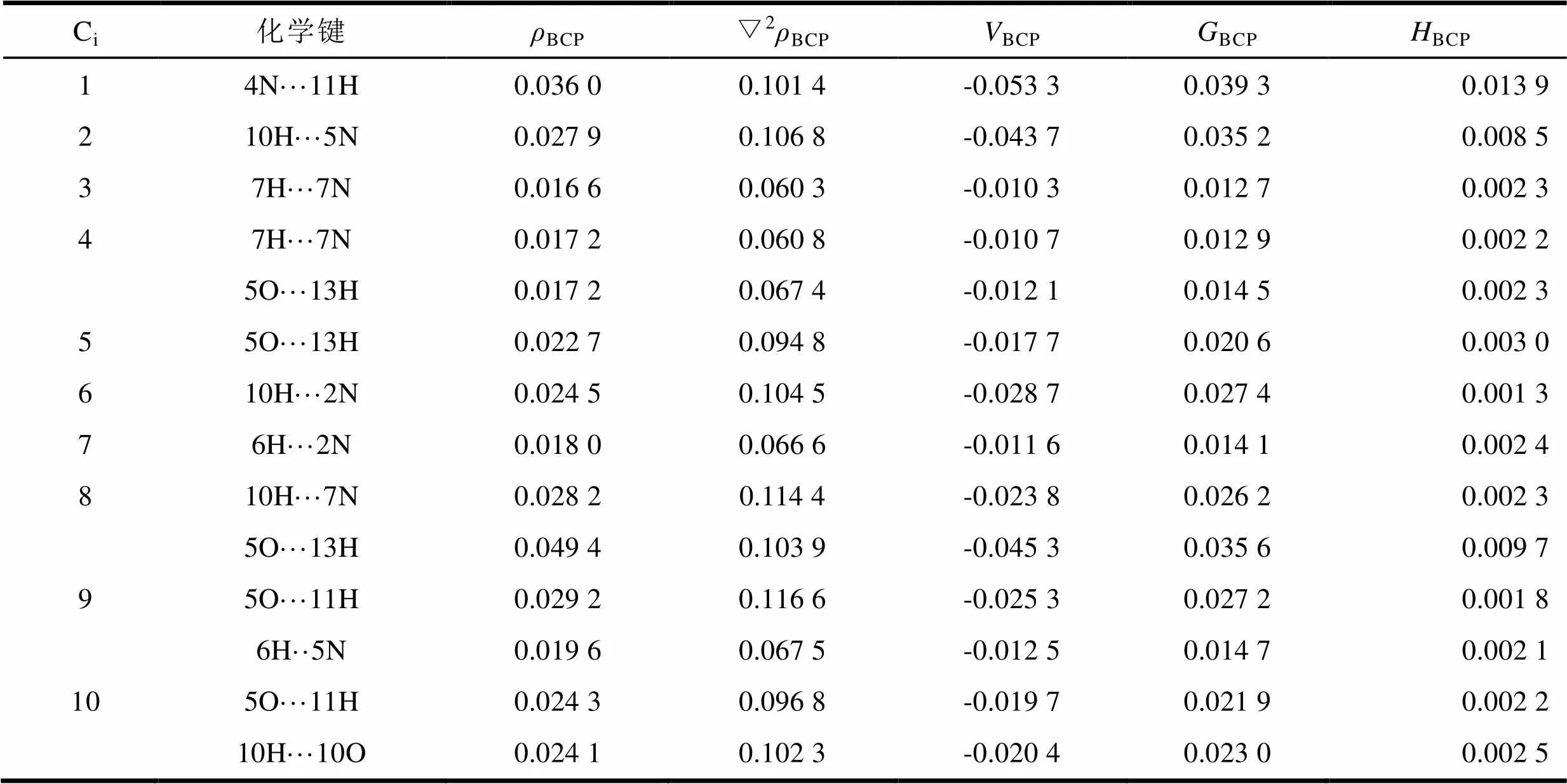
表3 甘氨酸-6-羟基嘌呤复合物主要临界点的电子密度拓扑参数(单位:a.u.)
4 结论
采用密度泛函理论在M602X/6-311+G(d,p)基组水平上对甘氨酸、6-羟基嘌呤、甘氨酸-6-羟基嘌呤复合物进行了结构优化和频率计算,得到了10种稳定的构型,其中单氢键复合物6种,二氢键复合物4种。对这些复合物经过计算、NBO、AIM分析后得到以下结论:
(1)根据相互作用能分析,二氢键复合物的稳定性高于单氢键复合物,分子中的氢键数目越多,复合物越稳定,二氢键复合物8在所有复合物中最稳定;
(2)对振动频率进行分析,得到复合物所形成的氢键都是正常的红移氢键;
(3)NBO分析结果表明,相关的氢键的(2)均为正值,证明复合物间存在氢键作用;
(4)AIM分析结果表明,所得的复合物的相互作用均为闭壳层体系之间的氢键弱相互作用,二氢键复合物8是复合物中成键临界点电荷密度最大的,相互作用能最大的,分别为0.038 8 a.u.、-70.56 kJ·mol-1,氢键相互作用最强,是最稳定的复合物。
[1] Greve C, Preketes K, Fidder H, et al. N-H Stretching Excitations in Adenosine- Thymidine Base Pairs in Solution: Pair Geometries Infrared Line Shapes and Ultrafast Vibrational Dynamics[J]. The Journal of Physical Chemistry A, 2013, 117(3): 594-606.
[2] Zheng Y Z, Zhou Y, Liang Q, et al. A theoretical study on the hydrogen- bonding interactions between flavon- oids and ethanol/water[J]. J Mol Model, 2016, (22): 95.
[3] 林雪飞,孙成科.6-烷基鸟嘌呤与DNA碱基间氢键作用的理论研究[J].化学学报,2016,68(16):1553-1560.
[4] 陈自然,何展荣,张宇红,等.C6H5O-H…X分子间氢键的理论研究[J].四川师范大学学报,2013,36(3):435-439.
[5] 蔡皖飞,郑妍,李来才,等.儿茶素-腺嘌呤分子间相互作用的密度泛函研究[J].四川师范大学学报(自然科学版),2014,37(1):115-121.
[6] 向铮,吴秀,郑妍,等.木犀草素与胞嘧啶相互作用的理论研究[J].化学学报,2011,69(17):1980-1986.
[7] 裴玲.肾上腺素与胞嘧啶相互作用的密度泛函理论研究[J].分子科学学报,2018,34(1):64-69.
[8] 和芹.甘氨酸与6-硫嘌呤氢键作用的密度泛函理论研究[J].南开大学学报(自然科学版),2009,42(1):16-21.
[9] 石云,朱敏.甘氨酸和水(1:1)氢键相互作用的理论研究[J].临沂师范学院学报,2009,31(3):71-75.
[10] 林江华,王秀丽,宋来洲.毒死蜱与DNA嘌呤碱基作用机制的研究[J].唐山师范学院学报,2017,39(5):3-8.
[11] 裴玲.没食子酸丙酯抗氧化活性及机理的理论研究[J].唐山师范学院学报,2016,35(5):31-34.
[12] Nikolaos N, Dimitrios S. Radical scavenging activity characterization of synthetic isochroman-derivatives of hydroxytyrosol: A gas-phase DFT approach[J]. Food Research International, 2015, (76): 506-510.
[13] V. Deephaa, Praveena, K. Sadasivam. DFT studies on antioxidant mechanisms, electronic properties, spectro-scopic (FT-IR and UV) and NBO analysis of C-glycosyl flavone, an isoorientin[J]. Journal of Molecular Structure, 2015, (1082): 131-142.
[14] Wang G R, Xue S H, An L, et al. Theoretical study on the structural and antioxidant properties of some recently synthesised 2, 4, 5-trimethoxy chalcones[J]. Food Chemistry, 2015, 171: 89-97.
[15] J. P. Foster, F. Weinhold. Natural Hybrid Orbitals[J]. Am Chem SOC. 1980, 102: 7211-7218.
[16] Bader W F. Atoms in Molecules a Quantum Theory[M]. Oxford: Oxford University Press, 1990.
[17] Popelier L. Atoms in Molecules a Quantum Theory[M]. Oxford: Oxford University Press, 1998.
[18] Krzysztof S and Bogumil J. Comment on “On the importance of the fragment relaxation energy terms in the estimation of the basis set superposition error correction to the intermolecular interaction energy”[J]. J Chem Phy, 1998, 109: 1198-1200.
[19] Frisch M J, Trucks G W, Schlegel H B, et al. Gaussian 09, Revision D.01[CP]. Gaussian, Inc.: Wallingford, CT, 2009.
[20] Lu T, Chen F. Multiwfn: A multifunctional wave- function analyzer[J]. Comput Chem, 2012, 33(5): 33- 580.
[21] Mohajeri A, Nobandegani F F. Detection and evaluation of hydrogen bond strength in nucleic acid base pairs[J]. J Phys Chem, 2008, (A112): 281-295.
[22] Hobza P, Havlas Z. Blue-shifing hydrogen bonds[J]. Chem Rev, 2000, 100: 4253-4264.
Theoretical Study on the Hydrogen-bonding Interactions between Glycine and 6-hydroxypurine
PEI Ling, HAO Wei
(Department of Chemical Engineering and Safety, Binzhou University, Binzhou 256603, China)
The structures and frequencies of glycine, 6-hydroxypurine and their complexes were optimized and calculated using the M602X method with the 6-311+G(d, p) basis set of the density functional theory. 10 stable glycine- 6-hydroxypurine complexes were found. The interaction energies of all the complexes with basis set superposition error (BSSE) correction are -14.60~-70.56 kJ·mol-1, which are in line with the energy range of hydrogen bond. The interaction energies are mainly contributed by hydrogen bonds, and related to the number of hydrogen bonds. Hydrogen bonding properties and characteristics of 10 compounds were analyzed with theories of natural bond orbital (NBO) and atoms in molecules (AIM). The topological analysis of the electron density shows that Y···H has a bond saddle point, and both the electron density and the Laplacian value fall in the hydrogen bond range. The interactions of the complexes are all closed shell systems.
density functional theory (DFT); hydrogen bond; natural bond orbital (NBO); atoms in molecules (AIM)
O641.3
A
1009-9115(2018)06-0036-05
10.3969/j.issn.1009-9115.2018.06.008
山东省自然科学基金联合专项计划项目(ZR2015BL012)
2018-07-22
2018-10-15
裴玲(1980-),女,山东邹平人,硕士研究生,讲师,研究方向为量子化学计算。
(责任编辑、校对:琚行松)
