THRβ基因R383H点突变致甲状腺激素抵抗综合征1例
2018-12-20邵飞张晓文沈山梅祝林迮雨阳毕艳
邵飞 张晓文 沈山梅 祝林 迮雨阳 毕艳
患者女性,61岁。因“甲状腺腺瘤术后6年,怕冷、乏力6个月余”于2017年2月5日入住南京大学医学院附属鼓楼医院内分泌科。6年前患者体检发现甲状腺双叶肿块,当时无怕热、多汗,无心悸、烦躁,无声音嘶哑,就诊于南京大学医学院附属鼓楼医院甲乳外科,行甲状腺彩超检查提示双侧甲状腺占位,左侧大小为3.0cm×2.0cm,右侧大小为1.0cm×1.0cm,甲状腺功能检查:血清促甲状腺激素(TSH)4.0mIU/L(正常参考范围 0.27~4.2mIU/L), 血 清 游 离 甲 状 腺 素(FT4)26.16pmol/L(正常参考范围12~22pmol/L),血清游离三碘甲状腺原氨酸(FT3)7.91pmol/L(正常参考范围 3.1~6.8pmol/L),垂体MRI检查提示垂体微腺瘤,考虑无功能性腺瘤。患者于2010年11月10日行甲状腺双叶次全切除术,术后病理提示结节性甲状腺肿伴腺瘤样结构。术后定期复查甲状腺功能,并给予左甲状腺素钠片替代治疗。患者于2016年7月出现怕冷、乏力、头晕等不适,尽管多次在南京大学医学院附属鼓楼医院门诊调整药物,但怕冷、乏力等症状未得到明显改善。家族中无类似病史。否认颈部放射线接触史。
本次入院体格检查:身高168cm,体重 55kg,体温 36.5℃,血压 143/82mmHg(1mmHg=0.133kPa),心率 77次 /min,呼吸18次 /min,BMI 19.5kg/m2,腰臀比(WHR)0.787。发育正常,查体合作。无眼球突出,活动自如,眼征阴性。颈静脉无充盈,气管居中,颈部可见一长约3cm手术瘢痕,已愈合。甲状腺无肿大,未扪及结节,未闻及血管杂音,指颤征阴性。心肺、腹部未查及阳性体征,四肢肌力正常,双下肢胫前无水肿。辅助检查:血常规、粪常规及尿常规正常;甲状腺功能:TSH 16.94 mU/L,FT426.64 pmol/L,FT35.43 pmol/L;抗甲状腺球蛋白抗体(TgAb)22.52U/ml(正常参考范围0~115 IU/ml),抗甲状腺过氧化物酶抗体(TPOAb)27.74U/ml(正常参考范围0~34U/ml);血生化正常;皮质醇节律正常;垂体MRI检查提示微腺瘤;心电图提示:室性期前收缩,房性期前收缩,短阵房速。基因检测:外周全血送至北京迈基诺医学检验所检测。结果提示:THRβ基因出现1个杂合子突变,编码区第1148号核苷酸有鸟嘌呤变为腺嘌呤,导致第383号氨基酸由精氨酸变异为组氨酸,为错义突变(如图 1)。
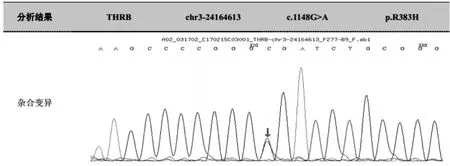
图1 THRβ基因测序结果
讨论甲状腺激素抵抗综合征(RTH)是一种以对目标组织具有不同程度的甲状腺激素敏感性减弱为特征的一种疾病。RTH是甲状腺功能障碍的罕见原因,临床上容易造成误诊和漏诊,因此有必要对罕见病例总结分析,降低误诊、漏诊的发生。
RTH是一种罕见的家族遗传性疾病,以常染色体隐性或显性方式出现,发病率约为1/50 000人。目前已经报道了THRα和THRβ两种形式的基因突变,不同突变形式可导致不同的临床表型。而临床上85%RTH是由编码TRβ的基因THRβ突变引起。RTH在临床上依据组织对甲状腺激素抵抗程度,分为全身型、垂体型及外周型。RTH患者可表现为无症状、甲状腺功能亢进或者甲状腺功能减退。因此,临床上容易引起对RTH漏诊与误诊。大多数RTH患者是无症状的,其自身可通过增加T4生成获得代偿作用,因而不需要医学干预。但少部分患者可能需要适当的治疗以改善甲状腺功能减退或甲状腺毒症症状。许多研究已经报道,使用包括抗甲状腺药物、溴隐亭、右旋甲状腺素(D-T4)、三碘甲状腺原氨酸类似物等多种药物来缓解甲状腺毒症或甲状腺功能减退症状[1-2]。然而,治疗效果却存在很大差异性。而目前还没有关于RTH治疗的明确指导方针。
本例患者为61岁女性,以怕冷、乏力就诊。患者6年前行“双侧甲状腺次全切术”,术后一直予以左旋甲状腺素钠片治疗。在治疗期间,经多次调整药物剂量,甲状腺激素水平仍未达标,并于2016年7月出现怕冷、乏力、头晕等不适。在治疗过程中,患者服用左旋甲状腺素钠片时,TSH水平始终保持在一个较高水平,而在患者改服甲状腺素片过后,TSH出现了下降趋势。当患者出现怕冷、乏力等不适时,再次给予适当剂量左旋甲状腺激素服用时,T3和T4保持较高水平,患者甲状腺功能减退症状没有改善,未出现甲状腺亢进症状。结合患者病史及临床表现,高度怀疑TSH瘤或RTH。该患者合并垂体微腺瘤,因此,判定垂体瘤是否分泌TSH尤为关键。除了影像学检查之外,促甲状腺激素释放激素(TRH)兴奋试验及生长抑素试验有助于两者的鉴别。在TRH兴奋试验中,血中TSH浓度迅速上升,20~30min达高峰,然后逐渐下降,120min时恢复正常,女性TSH反应稍高于男性。垂体TSH瘤患者中,TSH不能被兴奋,而RTH患者对TRH的刺激反应多为正常,峰值提前或者增高[3]。在生长抑素试验中,TSH瘤患者的TSH通常可被生长抑素抑制,甚至可使瘤体变小;而RTH患者对生长抑素的反应低下,其具体机制并不清楚。本例患者进一步基因测序发现THRβ基因出现R383H点突变,诊断为RTH。
此外,RTH在临床上还需要与罕见的家族性白蛋白异常性高甲状腺素血症相鉴别。血液中甲状腺激素的运输主要依靠与白蛋白的结合。家族性白蛋白异常性高甲状腺素血症则是白蛋白基因发生突变,导致白蛋白结构异常,使其与甲状腺激素亲和力增强,引起T4的升高[4]。但是患者的TSH、FT4通常正常。从TSH、FT4水平即可初步排除家族性白蛋白异常性高甲状腺素血症。而家族遗传倾向是两者的共同特征。值得注意的是,该患者于6年前行甲状腺双叶次全切,术后病理提示结节性甲状腺肿伴腺瘤样结构。既往研究表明TSH水平与甲状腺结节,尤其是甲状腺癌有着密切的联系,随着TSH水平升高,甲状腺结节的发生风险越大[5-6]。因此,不能排除患者的甲状腺腺瘤与长期的高TSH水平无关。从活检或者外科手术的RTH患者取得甲状腺组织,病理可见滤泡上皮有不同程度的增生,也有报道称腺瘤样甲状腺肿或者胶质样甲状腺肿,亦或是正常的甲状腺组织[7-8]。而在RTH患者中未报道合并有甲状腺癌的情况。该患者可能长期高TSH水平,致使甲状腺发生腺瘤样变。对于RTH合并甲状腺结节时,应明确结节性质,结合甲状腺彩超,必要时给予甲状腺细针穿刺,给予规范化诊疗。
既往对THRβ基因型R383H点突变仅有2篇文献报道。这2篇报道中患者临床症状均表现为甲状腺功能亢进,且均有家族性甲状腺疾病史。本病例未能采集到患者亲属的血样标本,不能详细了解该基因突变在其族系中变异情况。此外,本例患者与既往研究病例的临床表现截然相反,提示同一种基因突变,其临床表型并不一定完全相同,更说明基因诊断对于确诊本病的重要性。
在治疗方面,既往研究指出应该集中于改善患者的症状和临床表现,而不是旨在观察甲状腺激素水平正常化。大多数患者可通过增加自身甲状腺激素分泌来克服对甲状腺激素的不敏感,因此不需要治疗。对甲状腺功能亢进患者处理有些争议,不建议给予甲巯咪唑等抗甲状腺亢进药物治疗,应根据其主要症状给予β-受体阻滞剂或抗焦虑药物等症状治疗。对于TSH水平较高患者,3,3,5-三碘甲基乙酸(TRIAC)应用最为泛。TRIAC是三碘甲状腺原氨酸类似物,目前已被证明对儿童和成年人是有益的。
尽目前该病世界范围内已经报道了3 000多例,但仍然未能在治疗上达成统一共识。由于其罕见,临床上较多患者在确诊之前存在漏诊、误诊,部分患者甚至被给予了不恰当治疗。结合本例患者及既往报道,在高度怀疑RTH时,可以考虑优先选择基因检测,以便早期诊断。
