钙调神经磷酸酶Aβ基因沉默的乳鼠肥大心室肌细胞内向整流钾电流和动作电位的变化
2018-12-13扶泽南杨龙夏桂玲何炯红黄勇淇田野
扶泽南,杨龙,夏桂玲,何炯红,黄勇淇,田野
(1贵州省人民医院·贵州医科大学附属人民医院,贵阳550002)
心肌异常肥大导致的心室重构是慢性充血性心力衰竭(CHF)最主要的发生机制[1]。CHF患者最主要的死亡原因为心脏性猝死(SCD)[2]。心室肌细胞离子通道重构是导致细胞动作电位时程(APD)改变,进而引发恶性室性心律失常的重要病理生理基础[3,4]。内向整流钾电流(Ik1)参与动作电位复极后期进程,具有稳定心肌细胞静息电位、保障心脏复极储备的作用。研究[5,6]发现,编码Ik1离子通道α亚基的Kir2.1基因突变可导致长、短QT综合征。钙调神经磷酸酶(CaN)是迄今所知的惟一一种受Ca2+/钙调蛋白调节的丝氨酸/苏氨酸蛋白磷酸酶,广泛分布于机体各种组织中,参与多种信号转导通路,并通过作用于不同的底物而产生不同的生物学效应。CaN的活化可促进心肌肥厚性重构和心力衰竭,导致心律失常性死亡[7];CaN的活性改变影响肥大心肌细胞离子通道的重构[8~10]。目前关于钙调神经磷酸酶Aβ亚基(CnAβ)在CHF中的具体作用机制相关研究报道较少。2016年11月~2017年12月,我们观察了CnAβ基因沉默的乳鼠肥大心室肌细胞Ik1及动作电位的变化,旨在为CnAβ在CHF发生、发展中的作用机制提供理论依据。
1 材料与方法
1.1 实验动物、仪器与试剂 清洁级1 d龄SD乳鼠60只,雌雄不限,由北京大学医学部动物中心提供,许可证号[SYXK(京)2011-0039]。膜片钳放大器及数据转换器(美国Axon公司),兔抗大鼠CnAβ抗体(德国Merck Millipore公司),Ⅱ型胶原酶和胰酶(美国Sigma公司),高糖DMEM培养基、特优级胎牛血清(FBS)、苯肾上腺素(PE)和5-溴脱氧尿嘧啶核苷(5-BrdU)为美国Gibco公司生产,重组腺病毒shRNA干扰载体介导沉默编码CnAβ的基因(Ad-CnAβshRNA1,简写A1,5′-CAGAAAGGGTCTATGAAGCTTGTAT-3′)及腺病毒空载体由汉恒生物科技(上海)有限公司包装制备。
1.2 乳鼠心室肌细胞分离、培养及肥大刺激方法 取乳鼠,乙醚麻醉,开胸取心脏,分离心室并剪碎。酶解法分离细胞,差速贴壁结合5-BrdU培养获得心室肌细胞,37 ℃ 、5% CO2条件下培养48 h,换无血清DMEM高糖培养基。取对数生长期心室肌细胞,加入终浓度100 μM的PE刺激培养 48 h,RT-qPCR法测定细胞肌球蛋白重链β(β-MHC,上游引物序列5′-CAAGGAGCTCACCTACCAGA-3′,下游引物序列5′-CAGCTTGTTGACCTGGGACT-3′)、脑钠尿肽(BNP,上游引物序列5′-GTCTCCAGAACAATCCACGA-3′,下游引物序列:5′-CTAAAACAACCTCAGCCCGT-3′)[10]。结果发现,与刺激前比较,PE刺激48 h时心室肌细胞BNP、β-MHC的mRNA相对表达量均上调,说明PE可导致心室肌细胞肥大[10]。
1.3 心室肌细胞分组及CnAβ基因沉默方法 取心室肌细胞,分为A、B、C、D组。A组加入MOI=50对应量的腺病毒空载体,感染6 h时更换成2倍体积新鲜无血清DMEM,培养48 h。B组加入MOI=50对应量的腺病毒空载体,感染6 h换为2倍体积新鲜无血清DMEM,感染24 h时加入终浓度为100 μmol/L 的PE,培养48 h。C组加入MOI=50对应量的A1,感染6 h时换成2倍体积新鲜无血清DMEM,培养48 h。D组加入MOI=50对应量的A1,感染6 h时换成2倍体积新鲜无血清DMEM,感染24 h时加入终浓度为100 μmol/L 的PE,培养48 h。
1.4 各组心室肌细胞CnAβ蛋白检测方法 采用Western blotting法。取各组细胞检测心肌细胞CnAβ蛋白[10]。所有操作均严格按照试剂盒说明书进行。以目的条带的灰度值代表CnAβ蛋白的相对表达量,实验重复3次,取平均值。
1.5 各组心室肌细胞Ik1及动作电位变化观察 采用全细胞膜片钳实验。取各组细胞,在电压钳模式下记录各组Ik1,计算电流密度(pA/pF)=电流强度/电容。Ik1超极化阶跃方案:将钳制电压设置为-40 mV,-140 mV~-40 mV系列脉冲刺激,跃阶电压10 mV,波宽600 ms,频率0.1 Hz的刺激。电流钳模式下记录AP,给予1 nA电流脉冲,波宽2.5 ms,引发心室肌细胞AP。记录并分析动作电位复极20%、50%和90%(APD20、APD50、APD90)。实验重复3次,取平均值。
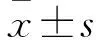
2 结果
2.1 各组心室肌细胞CnAβ蛋白相对表达量比较 A、B、C、D组心室肌细胞CnAβ蛋白相对表达量分别为1.0±0.01、2.59±0.30、1.00±0.16、0.82±0.16,与A组比较,B组心室肌细胞CnAβ蛋白相对表达量升高(P<0.05;n=3);与B组比较,C组心室肌细胞CnAβ蛋白相对表达量降低(P<0.05;n=3)。
2.2 各组心室肌细胞Ik1变化比较 各组心室肌细胞Ik1变化见表1,Ik1电流-电压图见图1。

图1 各组心室肌细胞Ik1电流-电压变化

组别电流密度-140 mV-130 mV-120 mV-110 mV-100 mV-90 mVA组-28.69±2.65-22.58±2.22-18.39±2.24-14.69±1.86-11.64±2.16-7.90±1.45B组-15.2±1.38a-12.4±0.86a-10.42±0.86a-8.48±0.74a-6.83±0.62a-4.92±0.46aC组-20.7±2.37b-16.8±2.13b-14.00±1.75b-11.20±1.59b-8.79±1.27b-6.36±1.04bD组-23.31±2.54-19.06±2.16-15.88±1.81-12.79±1.51-9.99±1.36-7.13±1.02
注:与A组比较,aP<0.05;与B组比较,bP<0.05。
2.3 各组心室肌细胞APD20、APD50、APD90比较 各组心室肌细胞APD20、APD50、APD90比较见表2。与A组比较,B组心室肌细胞APD20、APD50、APD90均升高(P均<0.05);与B组比较,C组B组心室肌细胞APD20、APD50、APD90均降低(P均<0.05)

表2 各组心室肌细胞APD20、APD50、APD90比较
注:与A组比较,aP<0.01;与B组比较,bP<0.01。
3 讨论
病理性心肌肥大是导致CHF的主要机制。恶性室性心律失常导致的SCD是CHF最主要的死因。Ik1在复极后期和稳定心肌细胞静息电位发挥着重要作用。在超级化状态下,内向电流显著增大;弱去极化时,电流改为外向,使膜电位保持在静息电位水平。当Ik1受到抑制时,可使膜电位升高,动作电位时程延长。慢性的血管紧张素Ⅱ刺激心脏能使Ik1电流密度减少,从而延长APD,导致LQTs[11]。心力衰竭时心肌细胞易发生晚期后除极(DADs)和触发活动,Ik1的下调为其主要机制之一[12]。在小鼠模型中,Kir2.1亚基过表达增加Ik1离子通道密度,保证快速和稳定的钾离子转运,稳定细胞膜电位,抑制折返性心动过速[13]。心力衰竭引发细胞水平的电生理重构,其中包括兰尼胆碱受体(RyRs)敏感性增加,促进钙离子释放和抑制Ik1;增加RyRs的敏感性和减小Ik1促进室性早搏和室性心动过速的发生[14]。有研究显示,α肾上腺素能受体激动剂甲氧胺干预能抑制兔心肌细胞的Ik1振幅[15];PE因导致Ik1减小而促进室性心律失常的发生[16];PE刺激单个分离的大鼠心室肌细胞15 min即导致Ik1的峰值减少45.8%[17]。
CaN参与心肌细胞肥大和心肌细胞膜多种离子通道结构和功能改变的调控。研究发现,CaN转基因小鼠发生显著的心室肥大,并进展为心室扩张和心力衰竭,给予CaN阻断剂环孢素A(CsA)或他克莫司(FK506)干预明显抑制该小鼠心室肥大,延缓心力衰竭的发生和减轻心力衰竭的严重程度[7]。给予血管紧张素II、PE或1%浓度的胎牛血清刺激培养的乳鼠心肌细胞,导致CaN活性增加、CnAβ基因和蛋白表达上调、心肌细胞肥大;给予CaN抑制蛋白cain/cabin 1和AKAP79干预能显著抑制该心肌细胞肥大和CnAβ基因及蛋白的表达[18]。在培养的乳鼠心房肌细胞,机械牵张刺激促进细胞肥大和CaNA亚基蛋白表达,抑制编码瞬时外向钾电流离子通道α亚基的Kv4.3蛋白表达;给予CsA干预,显著抑制牵张刺激诱导的心房肌细胞肥大、CaNA亚基蛋白表达上调和Kv4.3蛋白表达下调[9]。在培养的乳鼠心室肌细胞,给予PE刺激促进细胞肥大,上调细胞CnAβ蛋白表达,下调编码快钠电流离子通道α单位的Nav1.5 mRNA和蛋白表达;给予Ad-CnAβshRNA1沉默CnAβ基因干预显著抑制PE刺激的上述效应[10]。
CnAβ是CaN调控心肌肥大主要的功能结构。本研究采用CnAβ基因沉默的方法充分抑制其蛋白表达,使得CaN丧失发挥功能活性的物质基础,评估CaN活性充分抑制对Ik1的影响。结果显示,与未予PE刺激的null组细胞比较,在PE刺激的肥大心室肌细胞Ik1明显减小;而A1转染沉默CnAβ基因能够明显抑制PE诱导的心室肌细胞Ik1改变。
我们前期的研究结果显示,机械牵张刺激导致乳鼠心房肌细胞肥大,促进CaN A亚基和构成Ik1离子通道的Kir2.1蛋白表达,增大Ik1电流密度;CaN阻断剂CsA干预显著抑制牵张刺激的上述效应[8]。并且,我们在乳鼠心室肌细胞离体实验中发现,PE干预促成细胞肥大及CnAβ蛋白表达增加,调控组成钠离子通道的Nav1.5蛋白表达[10]。本研究中,PE刺激明显减小乳鼠心室肌细胞Ik1电流密度,延长APD20、APD50和APD90;敲低CnAβ基因显著抑制PE刺激对Ik1电流密度和APD的影响。机械牵张刺激和PE干预皆导致心肌细胞肥大和CaN A亚基蛋白表达增加,并且针对CaN抑制性干预皆有效,而两者Ik1电流密度的变化结果相反,原因不明。针对离体乳鼠心肌细胞Ik1的调控,不排除机械牵张刺激和PE干预存在CaN信号以外的作用机制影响干预效应,或者是因为心房肌和心室肌细胞Ik1离子通道存在差异所致。
综上所述,CnAβ基因沉默的乳鼠肥大心室肌细胞Ik1电流密度升高,心室肌细胞APD20、APD50、APD90均降低。CnAβ可能通过调节Ik1电流密度减小、APD延长参与心室肥大的发生发展。
猜你喜欢
——从一道浙江选考生物学试题谈起
