Illumina MiSeq高通量测序降解黑龙江绥化地区玉米秸秆细菌菌群的研究
2018-12-10,,,
, ,,
(1.哈尔滨商业大学食品工程学院,黑龙江哈尔滨 150076; 2.哈尔滨学院食品工程学院,黑龙江哈尔滨 150080; 3.哈尔滨学院食品生物技术重点实验室,黑龙江哈尔滨 150080; 4.哈尔滨工业大学环境学院,黑龙江哈尔滨 150090)
中国是农业生产大国,在农业生产中产生玉米等农作物的秸秆,由于秸秆降解速度慢、深加工附加值低等原因,造成秸秆相对过剩[1-3]。
我国黑龙江绥化地区是粮食作物的主要产区,但由于气温偏低,秸秆的微生物降解效率不高,焚烧的现象更为突出[4-5],成为秋冬季雾霾污染的主要原因之一,且存在安全隐患,因此,加强秸秆深度处理迫在眉睫[6-7]。
近年来,经过不断地研究开发,形成了以秸秆为原料的肥料化、燃料化、饲料化、原料化、基料化等途径,其中以物理化学处理方法为主,但处理产生的废弃物存在二次污染、成本昂贵且附加值低等问题[8-9]。利用微生物发酵分泌酶类来降解纤维素,具有条件温和、能耗低、无二次污染等特点,成为关注的焦点。目前,能够降解纤维素的微生物有很多,然而大多数研究都集中在常温、高温纤维素酶方面[10-11],关于低温纤维素酶的研究相对较少,所以对秸秆进行自然状态下低温高效降解细菌群落的研究,可以为我国黑龙江绥化地区秸秆处理方面提供有益的理论基础。
本研究采用MiSeq PE300多样性测序的方法对秸秆上细菌群落进行深入分析,可得到丰富准确的物种分类信息,具有数据准确、速度快、通量高等特点,并结合生信分析和16S rDNA基因预测等技术,对2015~2017年秋季秸秆样品进行采集和测序分析,获取秸秆上具有分泌纤维素酶功能的主要细菌群落的多样性和丰度信息,获得位于黑龙江绥化地区低温降解秸秆的主要细菌群落特征,为进一步生产低温降解秸秆的微生物菌剂提供理论依据。
1 材料与方法
1.1 材料与仪器
玉米秸秆 于2017年秋季采集黑龙江省明水县田间的玉米秸秆,将2015、2016、2017年玉米秸秆标记为A、B、C样品,采集完毕后将玉米秸秆用无菌剪刀、镊子剪成1~2 cm并用无菌袋封好,运回实验室后放入-80 ℃超低温冰箱中冻存,备用,样本信息详见表1;Bacterial Genomic DNA Extraction Kit 美国Biovision公司;QuickClean II PCR抽提试剂盒 南京金斯瑞生物科技有限公司;TransStart FastPfu DNA Polymerase 北京全式金微生物技术有限公司;TruSeqTMDNA Sample Prep Kit 美国Illumina公司。

表1 样品采样地点信息表Table 1 Sample sampling location information sheet
SW-CJ-2FD超净工作台 上海博迅实业有限公司医疗设备厂;BX-6便携式电热恒温培养箱 长春乐镤科技有限公司;HS-IE数显振荡培养箱 上海和盛仪器科技有限公司;Scan300全自动菌落计数仪 法国Interscience公司;DSX-280B手提式压力蒸汽灭菌锅 上海申安医疗器械厂;JJ224BC分析天平 常熟市双杰测试仪器厂;DW-86W100超低温冰箱 上海摩速科学器材有限公司;DYY-6C电泳仪 北京市六一仪器厂;GeneAmp9700 PCR仪 美国应用生物系统(ABI)公司;Miseq PE300二代高通量测序仪 美国Illumina公司。
1.2 实验方法
1.2.1 基因组提取 无菌状态下用镊子、剪刀、研钵对1~2 cm的秸秆样品进行预处理,称取A、B、C破碎样品各0.5 g,分别加入49.5 mL灭菌的生理盐水中,振荡混匀,吸取1 mL置入EP管中,3000 r/min离心20 s,吸取上清液,置入EP管,12000 r/min离心10 min,收集沉淀菌体,加入1 mL PE buffer混匀,按照Bacterial Genomic DNA Extraction Kit 基因组提取试剂盒说明提取三个样本基因组,平行3次,置于-80 ℃的超低温冰箱中储存待用。
1.2.2 琼脂糖凝胶电泳 将提取获得样品基因组进行1%琼脂糖凝胶电泳[12],条件为:电压5 V、30 min。
1.2.3 16S rDNA的PCR扩增 扩增秸秆基因组的16S rDNA片段,采用引物338F(5′-ACTCCTAC GGGAGGCAGCAG-3′)-806R(5′-GGACTACHV GGGTWTCTAAT-3′)扩增微生物基因组中16S rDNA的V4+V5区。PCR扩增条件为94 ℃反应4 min;94 ℃、45 s,55 ℃、45 s,72 ℃、60 s循环30次;72 ℃延伸10 min;4 ℃终止反应[13-14]。扩增产物长度预计436 bp。
1.2.4 PCR扩增产物琼脂糖凝胶电泳 扩增完成后,采用2%琼脂糖凝胶进行鉴定,检测各样品16S rDNA扩增产物。
1.2.5 Illumina MiSeq高通量测序 委托上海美吉生物医药科技有限公司 MiSeq建库,并进行生物多样性测序,测序后对DNA序列拼接和质控,并以此为基础进行生物信息学分析。
作为捷豹品牌的创始人威廉·里昂斯(William Lyons)爵士参与设计的最后一款捷豹轿车,于1968年巴黎车展首秀的XJ Series I是一款拥有媲美捷豹传奇跑车E-type驾控性能的豪华轿车。此外,XJ Series I还拥有大气沉稳、不失美感的外观造型以及优异的动力性能,为当时的豪华轿车市场注入了全新活力。当然,捷豹XJ自诞生之日起也赢得了众多车主的青睐,其中不乏社会精英、名流政要及皇室成员。
1.2.6 纤维素降解细菌菌株的筛选 在15 ℃温度下对玉米秸秆中低温降解纤维素菌株进行筛选,以玉米秸秆粉为唯一碳源进行富集,稀释涂布后反复分离纯化得到单菌落,用1 mg/mL刚果红染色液染色1 h,再用1 mol/L氯化钠溶液洗脱30 min,通过观察是否产生透明水解圈,初步判定所筛菌株对玉米秸秆的降解情况。玉米秸秆培养基、透明圈测定培养基等主要参照青格尔、姜立春等人的研究[15-16]。
1.3 数据统计分析
数据处理采用R语言和mothur(https://mothur.org/wiki/Main_Page)软件进行分析作图,测序获得基因序列与Silva(Release128 http://www.arb-silva.de)、EggNOG(evolutionary genealogy of genes:Non-supervised Orthologous Groups,http://eggnog. embl.de/)、KEGG(Kyoto Encyclopedia of Genes and Genomes,京都基因和基因组百科全书,http://www.genome.jp/kegg/)数据库进行比对。
2 结果与分析
2.1 琼脂糖凝胶电泳鉴定
由图1所示,B样本和C样本上微生物基因组电泳条带清晰,基因组提取效果良好,样本A上基因组电泳条带不清晰,需待PCR扩增后再次检测,以确认是否可用于后期实验。
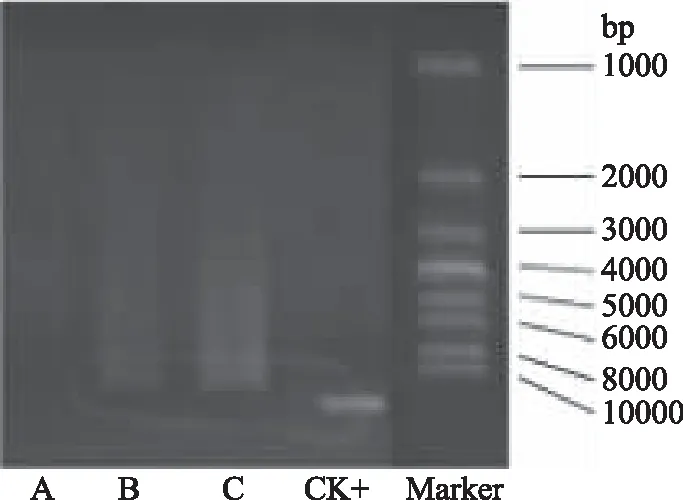
图1 秸秆基因组琼脂糖凝胶电泳图Fig.1 Straw genome agarose gel electrophoresis
由图2琼脂糖凝胶电泳结果可知,PCR扩增产物电泳条带位于400~500 bp之间,长度与预测一致,亮度适中,DNA含量较高,可继续进行实验。
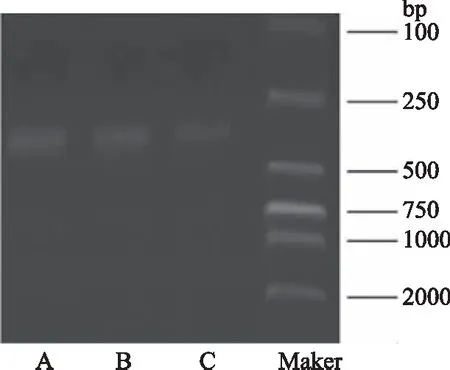
图2 秸秆细菌群落16S rDNA PCR扩增产物琼脂糖凝胶电泳图Fig.2 Agarose gel electrophoresis of amplified products of 16S rDNA PCR from straw bacterial community
2.2 测序数据初步分析
2.2.1 测序结果 对2015~2017年玉米秸秆中细菌群落进行测序,共获得61564385个碱基(base,bp),被聚类为582个OTU,共拼接成141219个16S rDNA基因片段,平均长度为430.06 bp。
2.2.2α-多样性分析 对测序结果进行α-多样性分析,以确定测序的质量,初步比较各样本细菌群落的多样性情况,见表2。
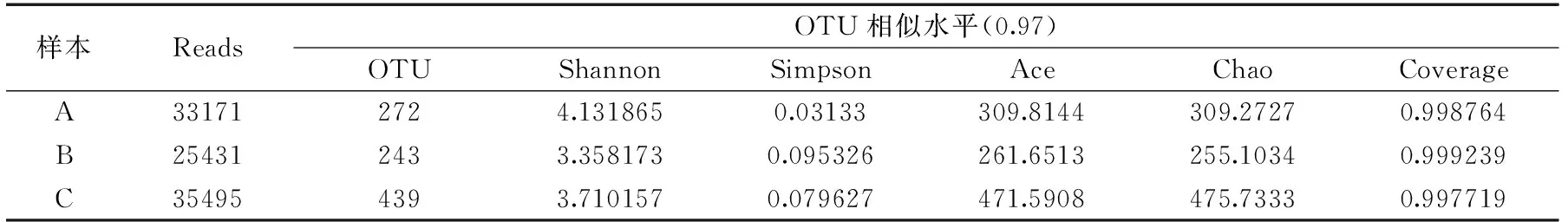
表2 样本Alpha多样性指数表Table 2 Alpha-diversity values of samples
由表2可见,C样本的OTU值、Ace值和Chao值远高于A样本和B样本;表明C样本中细菌群落丰富度较高;B和C样本的Simpson值高于A样本且接近,说明二者之间细菌群落的多样性相似,需进一步对比分析;各样本的Shannon值和Coverage值相差不大,说明三个样本之间具有相对独立性,且Coverage值较高表明此次测序覆盖率高,满足样本细菌多样性分析的需要,可进一步分析[17]。
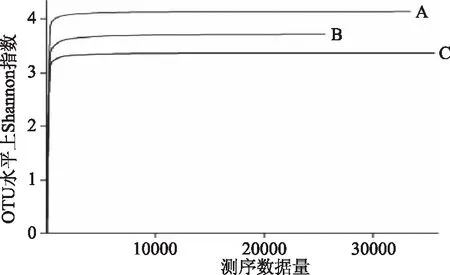
图3 秸秆细菌群落Shannon曲线Fig.3 Shannon curve of straw bacterial community
2.3 细菌群落组成分析
2.3.1 秸秆细菌群落组成 根据测序结果和Sliva数据库的比对结果,其中,Siwa数据库比对列14门、29纲、70目、136科、282属。对各样本细菌群落在属水平绘制累积图,见图4。

图4 秸秆细菌群落属水平组成分析Fig.4 Commposition analysis of the Genus level of the Straw bacterial community
研究发现,A样本物种丰度更加均匀,多样性更加突出,B样本次之。A样本中优势菌属主要为Rhizobium、Microbacterium、Sphingomonas、Devosia、Flavobacterium等;B样本主要为Pseudomonas、Streptomyces、Promicromonospora、Rhizobium等;C样本主要为Pseudomonas、Rahnella等。其中Pseudomonas、Devosia、Flavobacterium、Microbacterium、Streptomyces和Rhizobium等在秸秆中为主要优势菌属,且能够分泌纤维素酶,可作为低温降解秸秆的优先筛选菌。在后期研究中应重视分泌纤维素酶的优势菌群在不同温度、不同储存期间和不同营养状态的生存情况。
2.3.2 细菌群落Heatmap图 根据样本间丰度的相似性在属水平上进行聚类,对样本中丰度前30的细菌进行分析,得出细菌群落Heatmap图。通过颜色变化与相似程度来反映不同秸秆群落微生物在属水平上群落组成的相似性和差异性。

图5 秸秆细菌群落属水平Heatmap图Fig.5 Heatmap of the genus level of the straw bacterial community
Heatmap图清晰表现了各样本细菌群落的多样性和丰度,A样本和B样本多样性构成更加近似,在两样本中丰度高的属细菌主要为Pseudomonas、Streptomyces、Brachybacterium、Rhizobium、Devosia、Microbacterium和Flavobacterium等。丰度前30的细菌随着时间的推移产生一定的规律性,如:Sphingobium、Pseudomonas、Rahnella、Unclassfied_f_Micrococcaceae、Sphingomonas、Rahnella、Pedobacter等在时间延续的过程中,这些细菌对新鲜秸秆的降解能力较强,但随着时间的延长,丰度逐渐降低,生存能力下降,对秸秆纤维素的降解能力可能也会逐渐降低;Pseudoclavibacter、Stenotrophomonas、Altererythrobacter、Curtobacterium、Devosia、Brevundimonas、unclassified_o_Micrococcales、Rhizobium、Lysinimonas、Microbacterium随着时间的推移逐渐增加,丰度逐渐增加、活性逐渐增大,可能具有长期分泌纤维素酶来降解纤维素的能力。其中Pseudomonas、Rhizobium、Devosia、Microbacterium、Streptomyces、Flavobacterium等菌属一直存在且能分泌纤维素酶,是要研究的主要菌落,与累积图相符合。
2.4 细菌群落主成分分析
应用R语言进行PCA分析(Principal Component Analysis,主成分分析),分析OTU(97%相似水平)组成反应三年内秸秆的差异和细菌多样性,样本多样性组成越相似,在PCA分析图中的距离越接近。图6为秸秆细菌群落在OTU水平上的主成分分析图,PC1轴和PC2轴对样本组成差异的贡献值分别为69.28%和30.73%。通过分析发现,A、B、C样本间的距离均较大,细菌组成差异明显。图中A样本距离温度(T)、水分活度(Aw)远,说明T和Aw对新鲜秸秆内细菌群落多样性影响不大;B样本和C样本分别距离T和Aw向量的投影距离较小,即受影响相对较大,Aw越高,秸秆内细菌群落微生物生长越迅速,代谢活动也会随着温度的上升而增加,即随着储存时间的延长,秸秆内部的细菌多样性会随着T和Aw的变化而变化。
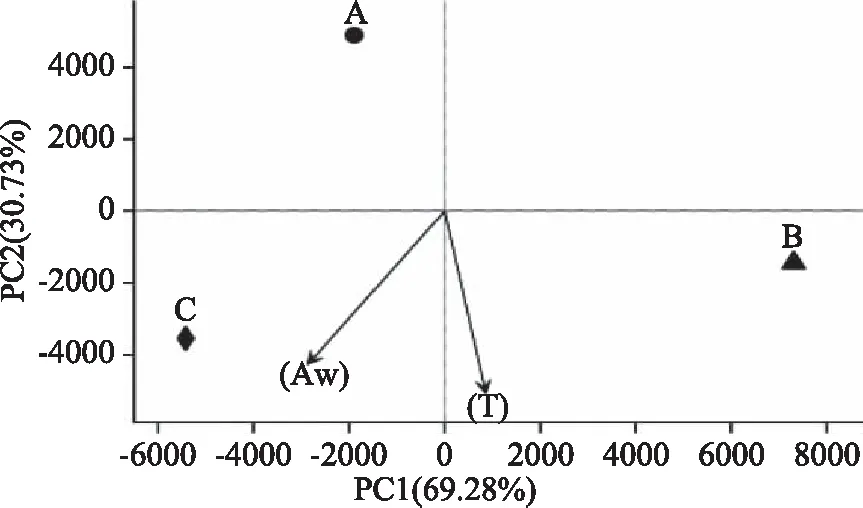
图6 秸秆细菌群落主成分分析Fig.6 Principal component analysis of the Straw bacterial community
2.5 样本中基因组COG功能分析
将测序获得的reads进行16S rDNA功能预测,比对EggNOG数据库,得到的COG功能分类统计图见图7。其中,横坐标为不同年份玉米秸秆,纵坐标为各样本中每个功能的丰度占该样品所有功能丰度的比值。
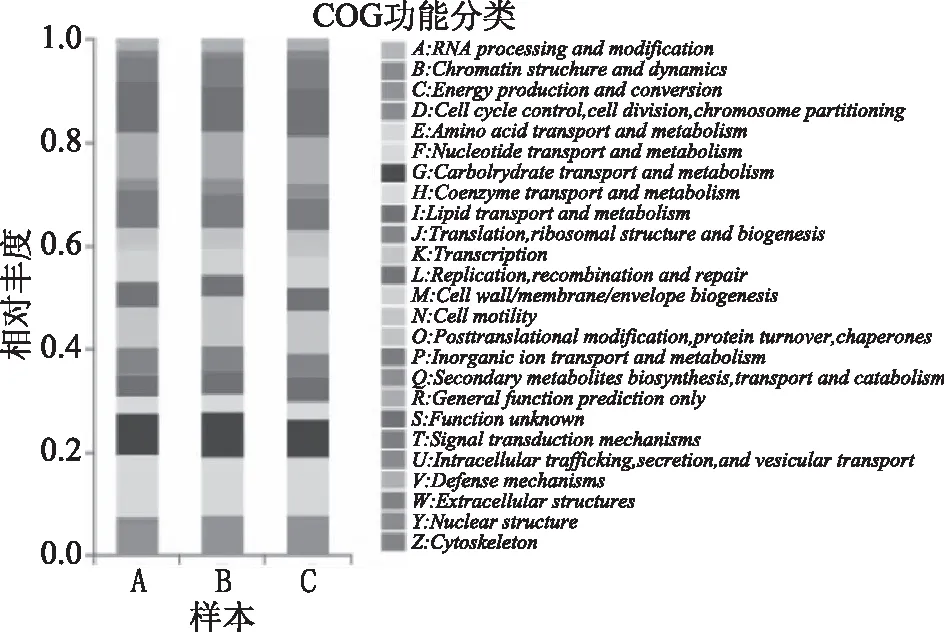
图7 秸秆细菌属水平上COG功能分类统计图Fig.7 Sample bacterial genera level COG function classification chart
从图中可以看出,与物种组成相比,样本A~C的COG功能组成相似,但丰度有所不同。Unigenes共有26个功能组,而在实验所采集的玉米秸秆样本中包含了22个功能组,从整体上看,B样本内微生物代谢功能更为丰富,其次为A样本。其中,B样本在能量产生与转化(C)、碳水化合物转运与代谢(G)、核糖体构成和生物合成(K)、次生代谢产物的生物合成/运输/分解代谢等功能上的丰度较高,可能与玉米秸秆分解代谢关系较紧密。
2.6 样本中基因组KEGG丰度统计与通路分析
通过测序获得的基因组与KEGG数据库进行比对[18],得到KEGG样品丰度统计表,如表3所示。
对不同年份玉米秸秆中基因组进行KEGG通路分析共获得241条代谢通路,表3中列出了7种与玉米秸秆在降解过程中主要分解代谢有关的代谢途径,其中ko00500代谢通路为秸秆中细菌菌群产生纤维素酶的主要通路。在ko00500代谢通路中,A样本中微生物的淀粉和蔗糖代谢功能明显高于B样本和C样本,说明随着储存时间的延长,秸秆中能够分泌纤维素酶的细菌菌群丰度逐渐增大,淀粉和蔗糖代谢能力逐渐增强。从整体上看A样本和B样本在玉米秸秆降解过程中的代谢通路功能较接近且高于C样本,与群落组成分析得到的结果一致。
2.7 降解纤维素功能的验证
研究从秸秆中筛选2.2.1中丰度较高且能代谢纤维素酶的两株细菌,在15 ℃温度下进行分离纯化,图8为Pseudomonas和Streptomyces分泌纤维素酶产生的水解透明圈。说明这两株细菌在低温下具有较强分泌纤维素酶的功能,与测序及预测结果一致,在后期的研究中,将主要对高丰度的细菌进行进一步的研究。
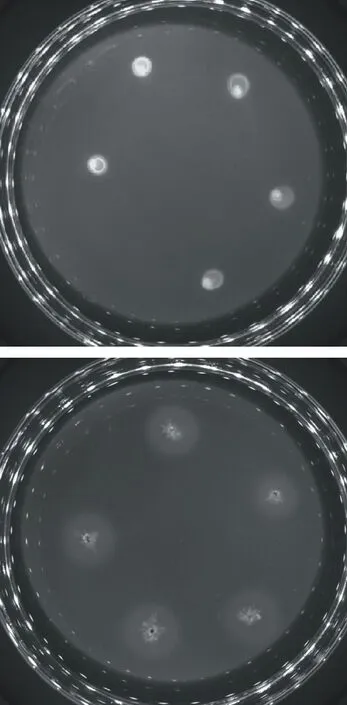
图8 秸秆细菌菌株刚果红培养基水解效果Fig.8 Hydrolytic effect of Congo red medium with bacterial strain of straw
3 结论
通过对玉米秸秆中细菌群落进行Illumina Miseq PE300多样性测序结果得知,2015年和2016年玉米秸秆细菌多样性高于2017年玉米秸秆,并得到以下结论:通过对样本内微生物群落结构与群落间的相似性、差异性比较分析,发现2015年玉米秸秆与2016年玉米秸秆细菌多样性及丰度较为接近;将测序得到的结果进行16S rDNA功能预测,通过COG、KEGG功能分析可知,2015年和2016年玉米秸秆中的微生物代谢功能较2017年玉米秸秆丰富;由分析结果可预测降解玉米秸秆的菌属主要有假单胞细菌属(Pseudomonas)、德沃西亚菌属(Devosia)、黄杆菌属(Flavobacterium)、微杆菌属(Microbacterium)、链霉菌属(Streptomyces)和根瘤菌属(Rhizobium)。综上,通过高通量测序技术可以全面掌握玉米秸秆内微生物的多样性和丰度变化,以及玉米秸秆降解过程中的代谢情况,为东北玉米秸秆生物资源化利用、寻找低温降解菌奠定一定的理论基础。
