对医疗器械监督抽验不符合标准规定产品企业进行分类处罚的研究
2018-12-06李沛李昕强小龙马凤莲吴植强
李沛,李昕,强小龙,马凤莲,吴植强
1 广西壮族自治区医疗器械检测中心,南宁市,530021
2 江苏省扬州市食品药品监督管理局,扬州市,225002
0 引言
2014年新修订的《医疗器械监督管理条例》(以下简称条例)颁布实施[1],新条例以分类管理为基础,以风险高低为依据,在完善分类管理、适当减少事前许可、加大生产经营企业和使用单位的责任、强化日常监管、完善法律责任等方面做出了较大修改。在法律责任方面,通过细化处罚、调整处罚幅度、增加处罚种类,增强了可操作性,加大了对严重违法行为的处罚力度。但对涉及条例的第六十六条情形(一)“生产、经营、使用不符合强制性标准或者不符合经注册或者备案的产品技术要求的医疗器械”[1]的处罚,与旧版条例比较,除金额外并没有实际性的改变。简单说来就是:凡是不符合标准规定的产品,不管是标识标签、说明书项目不符合还是其他安全、有效项目不符合,处罚的依据都是一样的。这有悖于本次条例修订的思想之一,即以风险治理为基础的分类管理。我们可以看到行政管理分类以风险程度大小为基础划分为三个类别管理;注册管理改为备案、省级注册和国家注册管理三种情况;生产和经营也按分级分类进行管理,处处考虑了风险的程度,但对涉及不符合标准规定产品的企业的处罚,却没有考虑其不符合项的风险大小是不同的。
1 现状
1.1 处罚措施单一
目前在食品药品监管部门网站上公开的对不符合医疗器械标准规定的产品企业处置方式都基本一致,就是罚款,如2016年所有的“北京市食品药品监督管理局关于国家医疗器械抽验不符合标准规定产品涉及企业处置情况的公告”,处置情况均为罚款。
国家食品药品监督管理总局发布的医疗器械质量公告中所有不符合标准规定的被检项目原来都是混合在一起发布的;2016年以后,质量公告中不符合标准规定的情况划分为“被抽验项目不符合标准规定的医疗器械产品”和“抽验项目为标识标签、说明书等项目不符合标准规定的医疗器械产品”两部分。这似乎在提示这两类不符合标准情况是有所区别的。但实际上,属地食品药品监督管理部门对相关企业进行调查处理时一般依据是按条例的第六十六条之情形(一)[1]。但该条款是不区分不符合标准规定的具体情况,导致处罚的手段基本一样,即罚款。要有区别仅在于在自由裁量权内的处罚金额大小和个别食药监局引用第六十七条之情形(二)进行处罚,即“生产、经营说明书、标签不符合本条例规定的医疗器械的”。然而笔者认为,引用第六十七条进行处罚,是适用条款错误。第六十七条之情形(二)所指“生产、经营说明书、标签不符合本条例规定的医疗器械的”,是指医疗器械说明书、标签不符合国家食品药品监督管理总局令第6号《医疗器械说明书和标签管理规定》[2]的情况,处罚不需要检验报告的支持。即使按第六十六条进行处罚,自由裁量的依据又在哪里呢?
1.2 风险分类治理思想未能体现在处罚措施上
产品被检项目的重要性是不一样的,或者说被检项目不符合标准规定时,其风险大小是不一样的。比如外观不符合标准规定与无菌不符合标准规定,外部标记不符合标准规定与漏电流不符合标准规定,其风险相差甚远,但处罚依据却都是一样,即按货值计算罚金。这显然不合理,也不科学,违背了过罚相当的基本原则。由于标识标签、说明书等项目不符合标准规定又比较容易发生,一方面给医疗器械检验检测机构带来了空前的压力,另一方面给社会和企业带来了过多的负面影响,群众认为我们的医疗器械质量堪忧,企业认为我们监管部门就是抠字眼。所以根据各检验项目的重要性进行分类处罚,势在必行。
2 对医疗器械产品质量特性重要度分级是分类处罚工作的基础
2.1 质量特性重要度分级在医疗器械生产质量管理规范中的作用
医疗器械本质上是工业产品,具有许多项质量特性。这些质量特性构成了产品质量的一切外在的特征和内在的特性,这些特征和特性的总和构成了产品的“适用性”[3]。但由于每一项质量特性的重要程度是不相同的,所以我们需要对这些质量特性重要度进行分级。质量特性重要度分级总体目的是设计开发的转换,即把产品的技术要求及设计重点具体地转告给工艺、制造、过程控制、物资采购部门和检验部门。具体体现在:
(1)可以在实现产品过程中方便确认产品的关键原材料和重要原材料,从而实现供应商和物料的重要性分类,这也是《医疗器械生产企业供应商审核指南》的要求。
(2)有助于完成不合格严重性分级的工作,便于综合评价产品质量,对提高检验工作质量、效率和减低检验费用有重要的意义。
(3)可以有效地识别产品生产的特殊过程、关键过程和重要过程,保证产品关键件、特殊件和重要件的质量,从而发挥质量综合管理和质量检验职能的重要作用[3]。
2.2 不合格严重性分级在医疗器械国家标准中的体现
质量特性既是医疗器械信息的基本内涵,也是诱发质量安全问题的内在因素[4]。不符合标准规定(不合格)是产品质量特性偏离规定要求的表现,而这种偏离因其质量特性的重要程度的不同和偏离规定程度的不同,对产品适用性造成的影响也不同[3]。一般医疗器械的国家标准将产品的不合格严重性划分为三个级别,分别用A、B、C表示,例如GB 15810—2001《一次性使用无菌注射器》的附录C,就对不合格严重性进行了分类。类似具有不合格严重性分级的医疗器械标准还有许多。
2.3 不合格严重性分级在部分医疗器械监督抽验检验方案中的应用
在既往国家医疗器械监督抽验的检验方案中,其实已经对某些产品的检验项目进行了不合格严重性的隐性分级。比如2015年的天然胶乳橡胶避孕套国家医疗器械监督抽验的检验方案,产品检验采用了GB/T 2828.4—2008《计数抽样检验程序 第4部分:声称质量水平的评定程序》的计数抽样检验方法,检验结果的判定原则(如13[0,1]、125[1,2]等),即计数抽样方案,就是根据不合格严重性的分级(不同的项目具有不同的AQL值)查询GB/T 2828.4—2008中的表1而来的;又如2017年的一次性使用无菌注射器监督抽验的检验方案也是暗含了不合格严重性的分级。
2.4 按风险进行分类处罚是科学监管的需要
遗憾的是,在对监督抽验检验结果后续处理工作中,完全忽略了质量特性重要度分级或者说不合格严重性分级这个问题。这固然有法规不完善的问题,如条例第六十六条内容粗放单一,与检验结果、标准的匹配度不够等问题,其实还有监管机构对质量管理不熟悉,不敢对处罚手段合理裁量的问题。
现代管理方法中需要运用抓“关键的少数”这一基本管理思想。少数关键质量特性对产品适用性有重要影响,即对产品功能、使用寿命和安全性有致命影响,如果偏离规定的要求,就会使产品严重丧失使用功能或危及人身安全;而大多数质量特性对产品的适用性只有较小的影响,其偏离规定的要求只是轻度影响、甚至不影响产品的主要技术性能,不会造成人身伤害[5]。质量特性重要度分级正是基于“不同的质量特性对产品适用性影响的重要程度不同”这一基本思想,用来追求以最小的经济投入获得最好的质量效益[5]。行政部门对于不符合标准规定的检验项目,采取一刀切的罚款处罚手段和措施,显然是合法不合理,也有悖于科学监管的思想。
3 以实例探索分类处罚的可行性
3.1 实例情况
2014年原食品药品监督管理总局发布了一期国家医疗器械质量公告(2014年第3期,总第4期),其中低中频治疗设备有25台产品部分被抽验项目不符合标准规定[6],不合格项目具体分布情况如表1。
3.2 按被检项目不符合标准规定的重要度划分的检验结果情况
从产品质量特性的重要程度、产品适用性的影响程度(安全性和有效性的影响程度)、顾客可能反映的不满意强烈程度等方面考虑,将不合格严重性进行分级,将不合格项目划分为A类、B类和C类,具体检验项目分类及不合格情况如表2。
3.3 分析
对质量公告附件进行统计分析后可以发现,有外部标记和/或随机文件(包括说明书)不合格的产品合计有21台;仅有上述项目不合格的产品有13台,占不合格产品总数的52%。这从总局历次发布的医疗器械质量公告中也可以看出, 外部标记、随机文件不符合标准规定的情况在不合格项目中的占比也是相当高。从表2可以看出,进行不合格严重性分级后,A、B、C类不合格产品的数量分别是4台、6台和22台,分别占不合格产品总数的16%、24%和88%。经统计分析,仅有C类不合格项目的产品有15台,占不合格产品总数的60%。
按照该质量公告,低中频治疗设备一共有25个产品被判抽验项目不符合标准规定,24家企业受到质量公告通报、罚款等措施的处理。如果按分类处罚的理念进行分类处置,假设C类不合格产品不上质量公告、不罚款,那质量公告中不符合标准规定的产品数就明显下降,占不合格产品总数60%的产品生产企业就有可能获得从轻处理,有着显著的社会意义。
3.4 讨论
对不符合标准规定的医疗器械企业进行分类处罚体现了以风险治理为基础的分类管理思想。可以从以下方面考虑实施:
(1)每年的国家医疗医疗器械监督品种确定后,由品种牵头单位组织生产、临床、检验和监管方面的专家对每个品种检验方案的检验项目进行不合格严重性分级,比如划分为A、B、C三类不合格项目。每年根据质量情况变化和监管需要对严重性分级微动态调整。
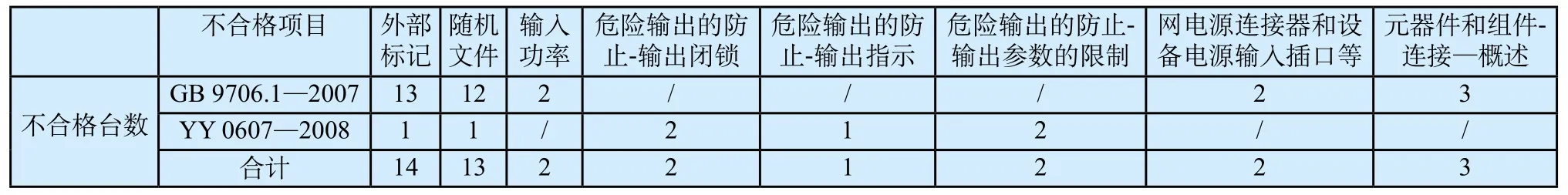
表1 低中频治疗设备不合格项目情况Tab.1 The unqualified items in low and medium frequency therapeutic instruments
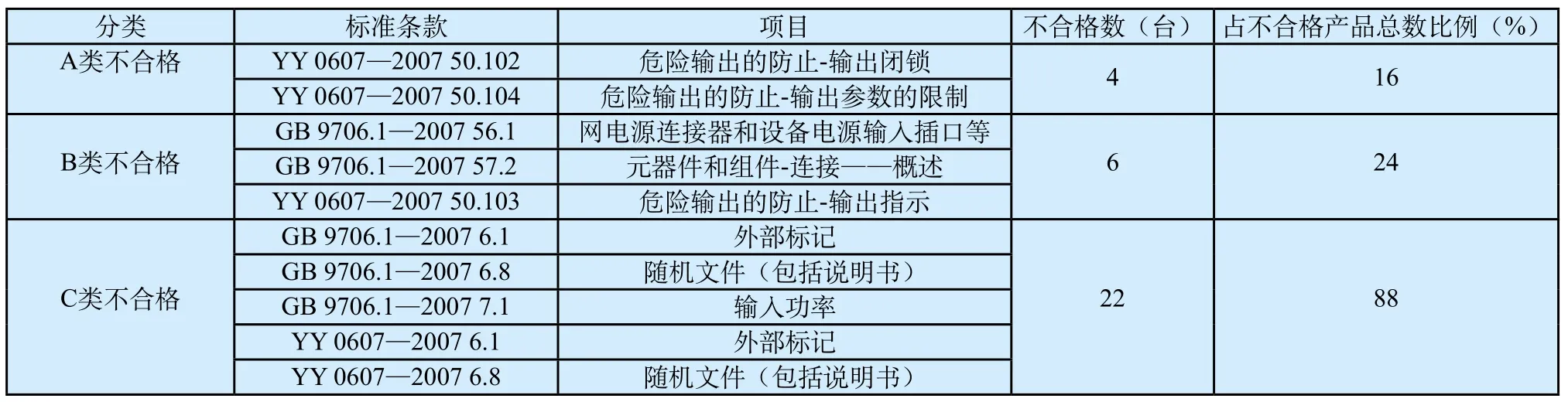
表2 按被检项目不符合标准规定的重要度划分的检验结果Tab.2 The inspection results classified according to importance degrees of the unqualified items
(2)监管部门按检验项目不合格严重性分类情况,制订不符合标准规定的分类处罚措施。目前相当多的省份药监部门已经编写有类似“行政处罚自由裁量权裁量基准”的文件,可在此基础上进一步细化要求。如出现C类项目不合格,可以先不上质量公告、不罚款,但勒令企业在规定时间内采取召回、整改等措施,如企业未按要求改正,则进一步加大处罚力度;B类项目不合格可采取罚款或上质量公告,召回、整改等措施;而A类不合格,则可以直接采取发布质量公告、罚款、召回、停产整顿、次年跟踪抽验等措施。
(3)进行不合格严重性分级讨论和确定,其意义还在于一方面在实施医疗器械生产质量管理过程中,有利于企业进一步确认产品质量特性重要度分级,有利于企业做好质量链中各环节的质量控制,有利于企业提高效率减低成本,有利于产业的发展;另一方面,各级监管人员不但有了重点监控产品的目录,还有了质量重点监控项目的目录,监管的目标更加明确,效率进一步提高。
4 结束语
《医疗器械监督管理条例》修订引入了风险治理为基础的分类管理思想,但在对不符合标准规定产品企业的处罚过程中,却没有考虑检验项目的不合格严重性程度或者风险大小不一的问题,千篇一律进行罚款处理,违背了立法的初衷,打击了生产企业加强质量管理的积极性。通过对不合格严重性进行分级,可以为现有法规下自由裁量处罚不符合标准规定产品企业提供一定的依据,避免过罚不相当的问题,也符合现代质量管理的理念,有利于企业持续提高和改进产品质量。
