Bcl-XL蛋白拮抗剂分子设计的研究进展
2018-12-04易享炎何天同张婉玲付豪亮汪丽泽杨雅琼
易享炎,何天同,曹 臻,张婉玲,付豪亮,汪丽泽,于 杨,黄 和,杨雅琼,黄 菲
(南京工业大学 药学院,江苏 南京 211800)
程序性的细胞死亡(细胞凋亡)在多细胞组织中具有重要的生物学功能,对于维持细胞存活与死亡之间的平衡至关重要。该平衡一旦被打破,将会出现严重的病理症状。目前研究发现:细胞过度凋亡与神经退行性疾病等相关,譬如阿尔茨海默病[1],凋亡受到抑制则会使存在DNA缺陷的细胞继续生长,导致肿瘤等疾病的发生。
细胞凋亡发生的生物学基础是蛋白质-蛋白质相互作用(protein-protein interactions,PPIs)[2],许多的生命活动都是依靠蛋白质间的相互结合或解离实现的[3]。B细胞淋巴瘤2(B-cell lymphoma 2,Bcl-2)[4]家族蛋白便是通过蛋白质-蛋白质相互作用产生效应的一类蛋白。Bcl-2家族蛋白是细胞级联凋亡路径上关键的效应因子,根据蛋白的具体功能,可分成促凋亡蛋白、抗凋亡蛋白和BH3-only蛋白3类[5-6](表1)。其中,BH3-only蛋白又被分为活化剂和敏化剂,活化剂可以直接激活促凋亡因子Bax、Bak[7-8],而敏化剂不能直接激活Bax、Bak,但可以抑制Bcl-2的抗凋亡功能。
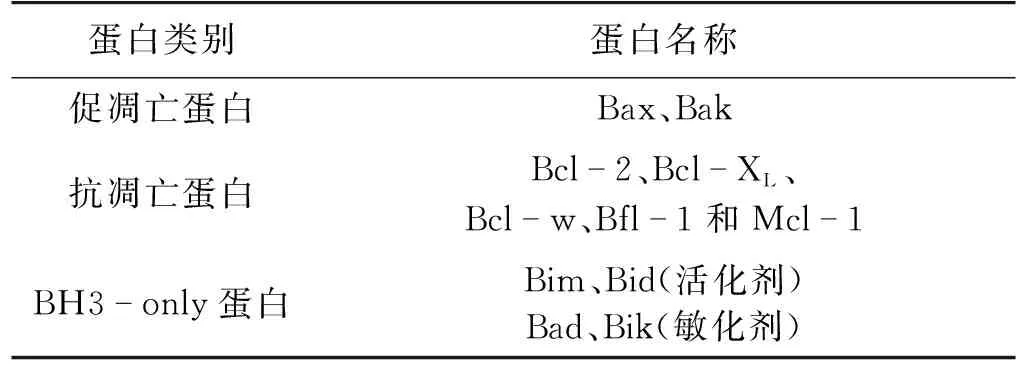
表1 Bcl-2家族蛋白成员分类
通常情况下,抗凋亡蛋白与促凋亡蛋白之间的平衡是细胞能否启动应激凋亡的主要因素。抗凋亡蛋白通过BH1、BH2和BH3结构域(图1)形成一个较长的疏水性沟槽来结合活化剂BH3-only蛋白上的BH3结构域,导致Bim和Bid无法激活Bax/Bak,阻止Bax/Bak寡聚化,并抑制Bax/Bak孔道的形成,继而不能触发细胞中线粒体外膜的透化(mitochondrial outer membrane permeabilization,MOMP)[9],也不能发挥有效的促凋亡活性;当受到外界凋亡信号刺激时,敏化剂BH3-only蛋白通过与抗凋亡蛋白结合,从而置换出活化剂BH3-only蛋白质,间接诱导Bax/Bak寡聚化,从而开启细胞凋亡程序[10-12](图1)。
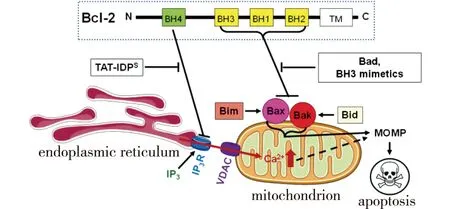
图1 Bcl-2家族蛋白诱导细胞凋亡机制[6]Fig.1 Mechanism of apoptosis induced by Bcl-2 family proteins[6]
在Bcl-2家族蛋白中,BH3结构域通常是促凋亡蛋白与BH3-only蛋白形成二聚体的必需结构。实际上,上述抗凋亡作用可以通过敏化剂Bad或BH3结构域的肽模拟物来抵消,开启细胞凋亡程序。同时,研究发现Bcl-XL、Bcl-2蛋白的过度表达是肿瘤发生及产生耐药性的重要原因[13]。
因此,设计出与Bcl-XL蛋白上的疏水沟槽有较高亲和力以及选择性的活性小分子已成为目前癌症治疗的热点研究方向之一。图2展示的是一种Bcl-2拮抗剂促凋亡的作用机制。
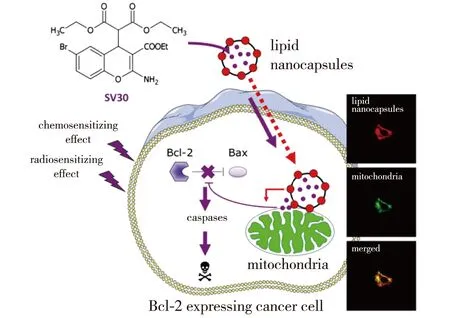
图2 Bcl-2拮抗剂SV30促细胞凋亡作用机制[14]Fig.2 The pro-apoptotic mechanism of Bcl-2 antagonist SV30[14]
使用脂质体纳米胶囊[14]对药物分子进行负载,将[2-氨基-6-溴-4-丙二酸二乙酯] -4H-色烯-3-羧酸甲酯SV30[15-16]运载到线粒体内膜后释放。该分子可以竞争性结合到Bcl-2蛋白上的疏水沟槽,从而阻断Bcl-2蛋白与Bax之间的联系,敏化癌细胞,开启细胞凋亡。
对目前现有的天然或人工修饰的抗肿瘤药物分子的结构进行分析,可以发现:具有苯甲酰基、氨基硫脲、硫代氨基甲酸酯、三氟甲基、二氟甲基、噻唑烷酮、苯并吡喃、三联苯和吡咯环等基团的化合物对诱导细胞凋亡具有一定的活性[14-16]。但对于复合结构的基团或骨架的研究,譬如本文中介绍的苯甲酰脲、磺酰胺和苯并噻唑这3类新型的Bcl-XL蛋白拮抗剂分子骨架,在国内的相关研究较少。笔者对上述Bcl-XL蛋白拮抗剂的发现历程、结构修饰以及活性测试3方面内容进行综述,并在此基础上对各类化合物对Bcl-XL蛋白的活性、选择性进行了相关评述。
1 具有苯甲酰脲骨架的Bcl-XL拮抗剂
2014年,Brady等[17]使用全新药物设计方法,将BH3结构域的疏水沟槽作为靶点[18-19],利用计算机辅助设计构建出以苯甲酰脲为骨架的BH3肽模拟物[20-21]。通过对该类化合物与Bcl-XL蛋白构效关系的研究以及X线单晶衍射实验的测定,揭示出一种新颖的缔合模型。
图3(a)为Brady等[17]选择苯甲酰脲作为基本骨架的构型。一方面由于该核心可以通过引入侧链基团向3个方向延伸,从而能够投射到沿着3个关键疏水氨基酸的侧链上,通过疏水作用力缔合。
另一方面,N—H会与羰基O形成稳定的分子内氢键作用,使该分子成为一个封闭构象核心,即使是在强极性和质子溶剂中也可以保持稳定,并且该氢键可以有效诱导顺式酰胺以及大环内酯的形成。图3(b)为P2、P3和P4疏水口袋,其对应的氨基酸分别是Leu151、Met154和Phe158[17]。
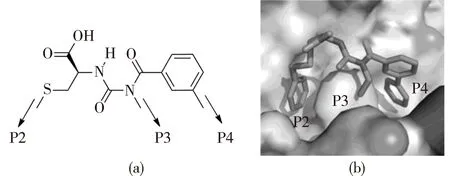
图3 苯甲酰脲骨架(a)和P2、P3和P4 3个疏水口袋(b)Fig.3 The skeleton of benzoyl urea (a) and P2,P3,P4 hydrophobic pockets (b)
根据3个疏水氨基酸残基的结构,Brady等[17]首先在苯甲酰环的间位引入芳基,将芳环投影到P4口袋中苯丙氨酸的残基上。由于结合在P3口袋中疏水性氨基酸残基是可变的,因此,在苯甲酰胺氮原子上取代了简单的烷基链。最后,为了模拟位于P2疏水口袋中异丙基的残基,在酰胺末端的碳上引入商业可购的S-苄基半胱氨酸。值得一提的是,羧酸基团在缔合过程中对静电相互作用是增加了疏水分子中极性成分的表面积(polar surface area,PSA)[17],使得该分子具有亲水性和亲脂性两重性(图4)。

图4 BH3结构域模拟肽化合物1Fig.4 The peptide mimetic of BH3 domain compound 1
使用邻近发光分析法(luminescence proximity assay,LPA)[22-23]评估化合物1与Bcl-XL蛋白之间的相互作用,发现化合物1对Bcl-XL蛋白的亲和性是相对较弱的,半抑制浓度值(IC50值)(指针对于Bcl-XL蛋白的半数抑制浓度)为128 μmol/L。因此,Brady等[17]依次对P2、P3和P4方向上的取代基进行筛选,发现P3方向的取代基可以分为芳基和烷基2类,芳基的IC50值均大于150 μmol/L,说明在该方向是芳基取代无活性;烷基取代基中尝试了正丙基、正丁基、异丙基和异丁基,发现只有当P3方向上氮上的氢原子被正丙基取代时2a,IC50值最低为109 μmol/L。
在保持P3方向氮原子上正丙基取代结构的同时,Brady等[17]还研究了P2方向上分子末端氨基酸部分的构效关系。一方面,亮氨酸衍生物2b基本上是没有活性的,表明芳环可能对该分子亲和力提升的贡献比较大;苯丙氨酸2c和酪氨酸2d衍生物也是没有活性的,说明末端芳环和酰胺羰基碳原子的间隔过近不利于亲和力的维持。另一方面,去除羧基基团2e会完全失去活性,当对羧基进行衍生化得到酯2f或酰胺2g时,都会导致活性降低。
上述分析证明了在P2方向上保留羧基结构的必要性,在此基础上Brady等[17]对氨基酸侧链进行筛选,发现当含有硫原子的异丁基取代时,2h的IC50值可降至15 μmol/L(表2)。

表2 P2方向上氨基酸种类对IC50值的影响[17]
Brady等[17]利用苄基、苯基乙炔、苯乙基和苯乙烯基在2h的基础上对其P4方向进一步衍生化,进行取代反应,发现苯基乙炔的效果最佳,IC50值为22 μmol/L,得到化合物3。利用卤族元素对末端苯环上的氢原子进行取代,得到化合物4,其IC50值最终降至13 μmol/L(图5)。

图5 BH3结构域模拟肽化合物3、4Fig.5 The peptide mimetic of BH3 domain compounds 3 and 4
对化合物3和4的X线单晶衍射进行分析,Lee等[24]发现这2种化合物与Bcl-XL蛋白的结合方式与最初设计的完全不同,原本放置在P4方向上的苯基乙炔基团,在实际的缔合模型中插入到了由P1和P2形成一个紧密的疏水口袋中,通过重叠模型可以看出:化合物3的苯乙炔基和ABT-737[24](Ki≤1 nmol/L,Ki:检测到50%抑制效果时,抑制剂采用Michaelis-Menten动力学计算获得的浓度)。联苯基在与Bcl-XL结合时空间取向是不同的,ABT-737的联苯基的空间取向是朝着P2方向的,从而证明了的确存在一种新颖的缔合模型,该化合物3、4与BH3疏水槽的结合方式跟先前发现的Bcl-2拮抗剂完全不同(图6)。

图6 化合物3、4与Bcl-XL疏水口袋缔合模型(a)和 (b)及化合物3与ABT-737重叠模型(c)Fig.6 Overall binding mode of compound 3 and 4 within the hydrophobic groove of Bcl-XL (a) and (b) overlay model of compound 3 and ABT-737 (c)
苯乙炔基在P1、P2中独特的取向可能有助于这类化合物对Bcl-XL蛋白的选择性。同时在与Bcl-XL蛋白形成的复合物中,2种化合物均通过分子内氢键来维持构象。由于苯甲酰脲环的假六元环的构象使其对Bcl-XL蛋白的疏水环境可能具有更好的选择性。Olaru等[25]利用表面等离子共振技术(surface plasmon resonance,SPR)对化合物3进行了3组平行实验,得到的平均值也反映出化合物3对Bcl-XL具有极高的亲和力,选择性较好(表3)。

表3 利用SPR对化合物3进行选择性测试
此外,对于上述化合物中的羧基部分似乎不与Bcl-XL蛋白发生相互作用,但消除羧基会使分子失去活性,所以羧基可能作用于识别Bcl-XL蛋白139号位精氨酸的残基。还可以发现:苯甲酰脲类化合物对Bcl-XL蛋白的抑制活性仍处于微摩尔水平,与已上市的模拟抑制剂ABT-737纳摩尔水平的活性相比,仍有较大的差距。因此,以苯甲酰脲为母核结构开发新型的Bcl-XL蛋白的拮抗剂,其抗肿瘤活性有待进一步的试验和确认。但Brady等[17]对通过对二者的空间缔合模型的研究,揭示出一种新颖的结合模式,将会对剖析Bcl-XL与Bcl-2的构象差异,针对Bcl-XL蛋白的靶向药物设计和开发具有指导作用。
2 具有磺酰胺骨架的Bcl-XL拮抗剂
Lock 等[26]在2005年通过基于核磁共振技术的活性化合物快速筛选方法,平行合成和基于结构的设计理念,发现了抗凋亡蛋白Bcl-2、Bcl-XL和Bcl-w的小分子抑制剂ABT-737,其亲和效力是之前已发现化合物的2~3倍。对于ABT-737作用机制的研究表明:ABT-737并不直接引发凋亡过程,而是发挥增强凋亡信号的作用,与化学疗法和放射疗法类似,会对正常细胞有杀灭作用,显示出协同的细胞毒性。ABT-737展现出最高的亲和力(Ki≤1 nmol/L),可以选择性地结合Bcl-XL、Bcl-2和Bcl-w;在人源淋巴瘤异种移植的小鼠模型中,单一给药疗效显著,目前正处于Ⅱ期临床研究阶段[26]。ABT-263是在ABT-737基础上进行改造,具有更好的药代动力学活性[26];ABT-199具有更加优良的选择性,已于2016年获FDA批准上市[27-29](图7)。

图7 具有促凋亡活性的BH3模拟物,ABT-737,ABT-263和ABT-199以及所提出的喹唑啉骨架Fig.7 Structures of BH3 mimetics with pro-apoptotic activity,ABT-737,ABT-263,ABT-199 and the proposed quinazolin scaffold
2.1 含有喹唑啉磺酰胺类的Bcl-XL拮抗剂
Sleebs等[30]根据生物等电子等排体的原理,设计出一系列含有喹唑啉结构的磺酰胺类化合物。在含有体积分数10%人血清的条件下,这类化合物对肺癌细胞系表现出纳摩尔水平活性。在对喹唑啉磺酰胺的化合物研究过程中,Sleebs等[30]发现其对Bcl-2家族蛋白的亲和力和选择性上存在一些差异,喹唑啉化合物对Bcl-2、Bcl-XL的亲和性明显大于其对Bcl-w,对Mcl-1几乎无活性。对比ABT-737和化合物5的结构可以发现,ABT-737中的苄基磺酰胺的结构被喹唑啉磺酰胺所替代。对化合物5(IC50值=3 nmol/L)与Bcl-XL所形成复合物的X线晶体结构进行分析,化合物5的结合模式类似于ABT-737,唯一的区别在于喹唑啉环上1号位的N原子会与Bcl-XL蛋白中101号酪氨酸羟基残基形成明显的氢键作用,见图8(b)虚线所示。
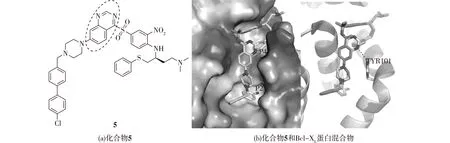
图8 化合物5与Bcl-XL蛋白复合物和(a)化合物5与ABT-737重叠模型(b)Fig.8 Compound 5overall binding mode of compounds 5 within the hydrophobic groove of Bcl-XL (a); overlay of compounds 5 and ABT-737 (b)
值得注意的是,虽然在ABT-737和Bcl-XL蛋白之间不存在额外的静电作用,但ABT-737对Bcl-XL蛋白的亲和力仍比化合物5高几倍。一般而言,对于含有喹唑啉结构的磺酰胺类化合物都是通过增强凋亡信号的机制来促使细胞凋亡,该类化合物是少数已知作用机制的Bcl-XL拮抗剂之一。
对于化合物5而言,虽然其在肺癌细胞系的活性测试中表现出纳摩尔水平,但相比于已上市的ABT-199仍有较大差距,其成药可能性较低。但值得注意的是,对喹唑啉环与Bcl-XL蛋白之间氢键作用的研究,将会对设计出特异性强的Bcl-XL蛋白拮抗剂具有一定的指导意义。
2.2 含有吡咯环磺酰胺类的Bcl-XL拮抗剂
2012年Zhou 等[31]设计并优化了一类含有4,5-二苯基-1H-吡咯-3-羧酸结构的Bcl-2和Bcl-XL的小分子抑制剂。其设计的基础是来自4,5-二苯基-1H-吡咯-3-羧酸这一新型具有药物样活性的骨架[32]。Aguilar 等[33]对化合物6,IC50值为(6±1) nmol/L与Bcl-XL蛋白的空间缔合模型的剖析为后续分子结构的优化提供了基础,报道了2种活性极高的化合物7,IC50值为(6.6±2.3) nmol/L和化合物8,IC50值为(4.8±0.1) nmol/L,对Bcl-2和Bcl-XL的Ki值均达到了纳摩尔级。
在药理活性功能评估中,上述2种化合物均被认定是Bcl-2和Bcl-XL的有效拮抗剂。此外,化合物7和8能以低至10 nmol/L的浓度诱导癌细胞迅速凋亡,并且发现化合物7可在人源肺癌细胞异种移植的动物模型中产生较强的抗肿瘤活性(图9)。
图9 化合物6(a)和化合物7(b)Fig.9 Compound 6 (a) and compound 7 (b)
Zhou等[31]通过对化合物6与Bcl-XL蛋白复合的晶体结构的分析,发现除了吡咯环外,噻吩环也可以与蛋白质形成疏水作用。同时,在吡咯环上N-甲基所处疏水口袋中,仍有容纳较大疏水基团的空间,因此设计了N-乙基(化合物7)、N-异丙基(化合物8)和N-环丙基,化合物7和8都表现出非常高地与Bcl-XL蛋白的亲和性(Ki<1 nmol/L)(图10)。

图10 化合物8Fig.10 Compound 8
研究还发现:化合物6虽然能够有效抑制肺部肿瘤的生长,但其在体内无法被完全的代谢,具有一定的细胞毒性。随后,Aguilar等[33]对吡咯环结构进行修饰,引入磺酰胺基得到化合物9。对化合物9在肺癌细胞模型的测试中发现,化合物9的Ki值小于1 nmol/L,IC50值为1~2 nmol/L。在已知对ABT-737、ABT-263敏感的3个肺癌细胞系中,化合物9可以有效地抑制肿瘤细胞的生长,在这些细胞系中,其活性是ABT-263的10倍,是ABT-737的50倍[33](表4)。化合物9能够在体内发挥快速、完整和持久的抗癌效果,是迄今为止报道的最有效的Bcl-2/Bcl-XL抑制剂,值得进行更加深入的评估,并可以作为潜在的临床抗肿瘤候选药物(图11)。

细胞系IC50值/(nmol·L-1)ABT737ABT263化合物9H196354.0±28.226.6±7.91.0±0.5H187137.7±71.338.4±26.81.4±1.3H1417173.4±122.154.2±11.12.3±0.2
3 具有苯并噻唑骨架的Bcl-XL拮抗剂
2014年Sleebs等[34]通过高通量筛选技术,筛选了100 000个结构不同的化合物,发现具有较强亲和性以及选择性的苯并噻唑腙类Bcl-XL蛋白的抑制剂。图12展示了以苯并噻唑腙为骨架的BcL-XL蛋白拮抗剂的演化历程,Sleebs等[34]对化合物10(IC50值=7.7 μmol/L)进行局部的化学修饰保留其苯并噻唑结构,同时保持腙键-芳香环周围2个可以自由旋转的构象与芳环之间的共面性,得到L-型化合物11(IC50值=0.12 μmol/L);引入亲脂性更好的四氢萘酮环得到化合物12(IC50值=0.013 μmol/L),化合物12对Bcl-XL蛋白的亲和力较化合物10提升了300倍;表面等离子体共振实验数据也进一步证实了化合物12对Bcl-XL疏水沟存在亲和力。WEHI-539由于其较差的物理化学性质,仅常作为工具分子使用。该系列化合物分子量很低(小于450),对Bcl-XL蛋白表现出极强的选择性,显示出潜在的成药性。
综上所述,研究者提供了一系列具有选择性和一定活性的Bcl-XL配体,并且指出:将化合物锁定在其结合构象中,可以潜在地增加与靶点结合的亲和力。该系列配体除了用于开发出调节Bcl-XL蛋白活性的新型药物外,对于阐明Bcl-XL在各类癌症中的参与以及生物学机制有着重要的作用。

注:MEF为小鼠胚胎成纤维细胞系;H146为人肺癌细胞系;EC50为针对小鼠胚胎成纤维细胞系的半数抑制浓度图12 苯并噻唑类化合物的衍生化Fig.12 Derivation of benzothiazole skeletal compounds
2014年,Tao等[35]发现含有苯并噻唑骨架的Bcl-XL蛋白拮抗剂,对于抗击慢性淋巴细胞性白血病(chronic lymphocytic leukemia,CLL)和非霍奇金淋巴瘤(non-hodgkin lymphoma,NHL)具有良好的治疗效果,从而利用计算机辅助药物设计以及X线单晶衍射技术,在保留起始化合物12亲和性和选择性的基础上,消除了其中腙的结构,最终得到化合物13(IC50值=0.091 μmol/L)。
化合物12虽然具有良好的亲和性及选择性,但是分子中含有“腙”这一结构。一方面,腙暴露于水中会释放有毒的2-肼基苯并噻唑;另一方面,化合物12及其类似物的药代动力学性能普遍较差,口服利用度很低。因此, Tao等[35]试图用水解稳定的化学键替代腙,同时保留分子其他有利的属性。应用分子对接技术将腙基核心结构转化为酰胺或脲基等基团,保留了与受体蛋白之间多种结合的相互作用。在大鼠给药实验中,化合物13这类重组骨架分子显示出比原始腙骨架更好的清除率和口服生物利用度。
Tao等[35]在这个理想的核心骨架基础上,通过在吡啶甲酸的3号位或噻唑环5号位引入芳环,可以与疏水性的P4口袋(图13)更好地缔合,从而提高了针对Bcl-XL蛋白的重组支架分子的生化特性和细胞亲和力。这种亲和力的改善也体现在小鼠胚胎成纤维细胞系(mouse embryonic fibroblasts,MEF)的活性测试实验中,化合物14的EC50值达到0.014 μmol/L,其对该细胞系增殖的抑制效果提高近20倍[36]。
此类小分子具有很强的选择性,为解决肿瘤治疗中耐药性的问题提供了新的研究方向,其亮点在于酰胺与腙结构之间的转化,这一思路将会为其他药物分子设计中,改善药代动力学活性提供借鉴。然而,在活性测试中,使用的细胞模型较为单一,因此该类小分子的抗癌活性有待于进一步的研究。

图13 化合物14(a)、15(b)以及在0.235 nm辨率下的观察到化合物15与Bcl-XL的相互作用(c)Fig.13 Compounds 14 (a), 15 (b) and interactions of compound 15 with Bcl-XL observed in a 0.235 nm resolution crystal structure (c)
4 结语
对苯甲酰脲骨架、磺酰胺骨架以及苯并噻唑骨架的Bcl-XL蛋白小分子拮抗剂的发现、衍生化过程及药物活性3方面进行了论述,重点评述了3类化合物分子设计的历程。发现苯甲酰脲作为一种新型骨架模型,对于Bcl-2家族蛋白具有一定的活性,但对Bcl-XL蛋白的选择性尤为突出,该骨架模型的发现,揭示了一种新型的缔合模型,对于以Bcl-XL蛋白为靶点的药物分子设计具有极强的指导意义。以磺酰胺为核心骨架的Bcl-XL蛋白拮抗剂,是目前研究最为充分的一类基于蛋白质-蛋白质相互作用小分子抑制剂,其中部分化合物已经成药。对磺酰胺为核心的骨架进行修饰,得到一系列亲和活性优良且选择性较好的化合物,例如化合物5和9,丰富了此类小分子拮抗剂的种类,为其进一步成药奠定基础。在以苯并噻唑为骨架的这类化合物中,利用酰胺和脲结构替换腙骨架,从而获得较好口服生物利用度,成为潜在的抗肿瘤药物候选化合物。
3类化合物分别代表着现代抗肿瘤药物设计的3个发展方向,从改善药物分子选择性到提升药物分子的活性,再到提高药物的生物利用度。相信随着人们对药物设计理念和方法的不断创新以及小分子药物文库的不断丰富,会有更多Bcl-XL蛋白拮抗剂被开发出来。
