Cu掺杂6000系铝合金中βʺ相的第一性原理研究
2018-11-17温柏杨贾志宏吴小志
温柏杨,贾志宏, 2,吴小志,刘 庆
Cu掺杂6000系铝合金中βʺ相的第一性原理研究
温柏杨1,贾志宏1, 2,吴小志3,刘 庆1
(1. 重庆大学 材料科学与工程学院,重庆 400044;2. 重庆大学 电子显微中心,重庆 400044;3. 重庆大学 物理学院,重庆 400044)
采用基于密度泛函理论的投影缀加平面波方法和广义梯度近似,研究Cu掺杂对6000系铝合金中主要强化相ʺ相(Mg5Al2Si4)的几何结构、相稳定性和电子结构的影响。结果表明:ʺ相的晶胞参数与文献报道相符。掺杂Cu后体系的晶胞形状发生微小变形且体积减小,而不同掺杂浓度和掺杂位置对掺Cu结构Mg5−xAl2−ySi4Cu+y的几何性质影响不同,进而影响ʺ相和Al基体之间的晶格错配度;Cu既替代Mg1又替代Al原子和Cu只替代Al原子的结构在合金中更容易形成,而Cu只替代Mg1原子的结构在合金中不易形成,该计算结果与实验报道相符。电子结构分析表明,掺杂Cu后形成的Mg5−xAl2−ySi4Cu+y相结构的稳定性和体系在费米能级附近的赝能隙密切相关。
第一性原理;6000系铝合金;强化相ʺ相;相结构稳定性;Cu掺杂
6000系变形铝合金比强度高,成形性、焊接性和耐腐蚀性良好,大量用于建筑和汽车[1−2]等工业领域。ʺ相一般在6000系铝合金的峰值时效过程中析出,是影响6000系铝合金强度的主要强化相[3]。析出相的尺寸、密度、分布、本身的性质(析出相成分、结构、形状、稳定性等)以及与合金基体相互作用等是影响合金力学性能的主要因素[4−5]。加入合适的微量合金元素可以影响ʺ相的尺寸、密度、分布和本身的性质及与合金基体的相互作用,Cu是最常加入的微量合金元素。JAAFAR等[6]研究了Cu对6000系铝合金的组织影响,观察到添加微量Cu 可以使ʺ相的尺寸减小密度增加。MURAYAMA等[7]研究了Cu对6000系铝合金的时效硬化效果影响,发现微量Cu加入会增加ʺ相的密度进而影响时效硬度。Cu加入6000系铝合金后Cu元素对ʺ相的尺寸、密度和分布的影响已经有大量的研究[6−7],但关于Cu加入6000系铝合金后ʺ相的本身性质变化如几何结构、相稳定性等尚未见到系统的研究报道。虽然QIU等[8]采用分子动力学研究了Cu掺入对ʺ相的力学性能的影响,但对ʺ相的几何结 构、相稳定性和电子结构等的影响没有研究。ʺ相的几何结构变化会改变ʺ相与合金基体的相互作用,如ʺ相和6000系铝合金基体之间的错配。而ʺ的相稳定性变化会影响ʺ相的形成难易程度。ʺ相稳定性变化的内在原因是电子结构发生变化。所以研究Cu掺入ʺ相后ʺ相的几何结构、相稳定性和电子结构变化很有必要。
由于ʺ相的尺寸太小(4 mm×4 mm×50 nm)[9],用实验来表征Cu掺入对ʺ相的几何结构、相稳定性、电子结构等性质的影响比较困难,而材料的物理性质由其微观电子结构决定,原则上可以通过求解薛定谔方程求出材料的几乎所有的性质。基于密度泛函理论的第一性原理是用较少的近似方法和较少的实验数据求解薛定谔方程的方法,密度泛函理论的基本定理Hohenberg-Kohn定理指出从薛定谔方程得到的基态能量是电荷密度的唯一函数,且基态电荷密度唯一确定了基态的所有性质包括能量和波函数。所以采用第一性原理方法可以计算获得相关信息。为此,本文作者运用基于密度泛函理论的第一性原理计算方法研究了Cu原子进入ʺ相前后的几何结构、相稳定性和电子结构变化,并从电子结构的角度解释了Cu掺杂位置对ʺ相稳定性的影响机理。
1 理论模型与计算方法
ANDERSEN等[9]采用能谱仪(EDX)测得ʺ相中Mg、Si摩尔比约为0.8,认为ʺ相的成分为Mg5Si6。而在2009年[10],ANDERSEN等用原子探针层析技术(APT)和会聚束电子衍射研究ʺ相的成分和结构,结果表明,ʺ相中含有20%Al(摩尔分数),并且Mg、Si摩尔比约为1.1,Mg5Si6结构中的部分Si原子被Al原子替代。通过第一性原理计算表明,Al替代Si3原子后能量会降低,得出ʺ相的成分为Mg5Al2Si4时能量最低。WENNER 等[11]计算了不同成分ʺ相和Al基体的错配,其中Mg5Al2Si4与Al基体的错配与实验最为接近,进一步证实了ʺ相的成分为Mg5Al2Si4。所以本文的计算工作采用Mg5Al2Si4作为ʺ相的成分[12−13]。
Mg5Al2Si4的晶体结构呈单斜结构[9],如图1所示,空间群为2/(No.12),晶胞参数=1.516 nm,=0.405 nm,=0.674 nm,轴和轴夹角=105.3o,轴和轴夹角、轴和轴夹角均为90o。Mg5Al2Si4单胞中含有22个原子,6种非等效原子:2个Mg1、4个Mg2、4个Mg3、4个Si1、4个Si2和4个Al原子。
实验研究[13−15]表明,Cu会进入ʺ相的结构中替代其中的Mg1或Al原子。由于Cu可以替代Mg5Al2Si4单胞中的Mg1或Al原子[13−15],所以采用2个Cu取代Mg1, 2个Cu取代Al,则得到掺Cu的ʺ相晶胞模型,成分为Mg5−xAl2−ySi4Cu+y。掺入ʺ相的Cu摩尔分数(以下简称为(Cu))为(+)/11。由于Cu掺入Mg5Al2Si4有3种可能性:只替代Mg1原子、只替代Al原子、既替代Mg1又替代Al原子,本文选取计算模型时同时考虑了掺杂浓度和掺杂位置两个变量,最后选择了7个掺Cu的ʺ晶胞模型Mg5−xAl2−ySi4Cu+y进行计算,即(,)={(0.5, 0), (0, 0.5), (1, 0), (0.5, 0.5), (0, 1), (1, 1), (0, 2)},7个晶胞模型的掺杂浓度(Cu)={4.545, 9.09, 18.18}。
运用基于密度泛函理论(DFT)的第一性原理方法计算时,采用广义梯度近似(GGA)下的PBE (Perdew-Burke-Ernzerhof)[16]形式处理Mg、Si、Al和Cu的交换关联能,采用投影缀加平面波赝势 (PAW)[17−18]处理离子−电子之间的相互作用。计算中考虑的价电子构型分别为Mg 3s23p、Si 3s23p2、Al 3s23p1、Cu 3d104s1。用Monkhorst-Pack方法选取7×21×13的K点网格,500 eV截断能被用于所有计算,体系总能量收敛精度为1.0×10−5eV/atom,当体系平衡时,允许体系中每个原子所受的最大应力小于0.01 eV/Å。
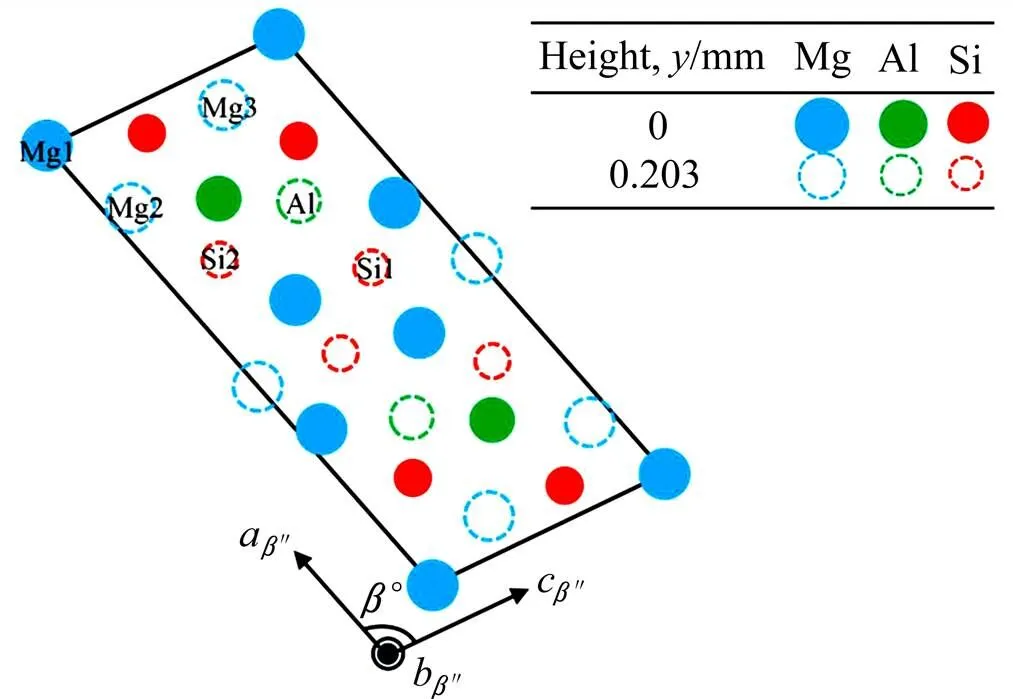
图1 Mg5Al2Si4的结构(绿色的为Al原子,蓝色的为Mg原子,红色的为Si原子)
2 结果与讨论
2.1 Cu的掺杂位置及掺杂浓度对掺Cu相几何性质的影响
为验证计算方法的合理性,首先计算未掺Cu的Mg5Al2Si4的晶胞参数,并与实验测得的数值[19−20]、第一性原理计算的结果[10]进行比较,结果见表1。由表1可以看出,计算所得的晶胞参数和体积与实验测得的数值非常接近[19−20],体积的误差不超过2%。同时,本文计算结果与HASTING等[10]计算的结果相比也非常接近。所以,未掺杂Cu的Mg5Al2Si4单胞的晶胞参数与文献报道相符,且本文的计算方法合理。
由于Cu掺入Mg5Al2Si4后,掺Cu的ʺ单胞Mg5−xAl2−ySi4Cu+y几何结构的变化在实验上和计算上尚未研究,所以本文计算了Cu替代Mg5Al2Si4中Al原子或Mg1原子后Mg5−xAl2−ySi4Cu+y的几何结构。掺Cu后的晶胞参数和体积见表2。由表2可知,掺入Cu后Mg5−xAl2−ySi4Cu+y的晶胞形状发生微小变形(晶胞的轴和轴夹角发生变化,变化范围为103.9°~107.2°)。
Cu在Mg5Al2Si4中有3种可能的掺杂位置:只替代Mg1,只替代Al,既替代Mg1又替代Al。掺Cu晶胞体积与掺杂位置、掺杂浓度的关系见图2。从图2可以看出,随着掺杂浓度的增大,不同掺杂位置的结构体积都逐渐减小。产生这种变化的原因是掺入原子Cu和被替代原子Mg1或Al的原子半径存在差异:Cu(0.128 nm)<Mg(0.160 nm),Cu(0.128 nm)<Al(0.143 nm)。比较图2 中相同掺杂浓度不同掺杂位置的结构体积,体积的大小顺序为:Al(只替代 Al)>Mg1-Al(既替代Mg1又替代Al)>Mg1(只替代Mg1)。该结果表明,虽然Cu掺入Mg5Al2Si4后3种掺杂位置的结构体积均降低,但在相同掺杂浓度的情况下,Al最大,Al-Mg1其次,Mg1最小,原因是Cu比Al和Mg都小,而且Al(0.143 nm)<Mg(0.160 nm)。

表1 Mg5Al2Si4的晶胞参数和体积
Change rate of volume:/0=(0−)/0, where0represents Mg5Al2Si4volume obtained by calculation of this work,represents Mg5Al2Si4volume obtained by experiments[19−20].
表2 掺Cu结构Mg5−xAl2−ySi4Cux+y的晶胞参数β和体积V
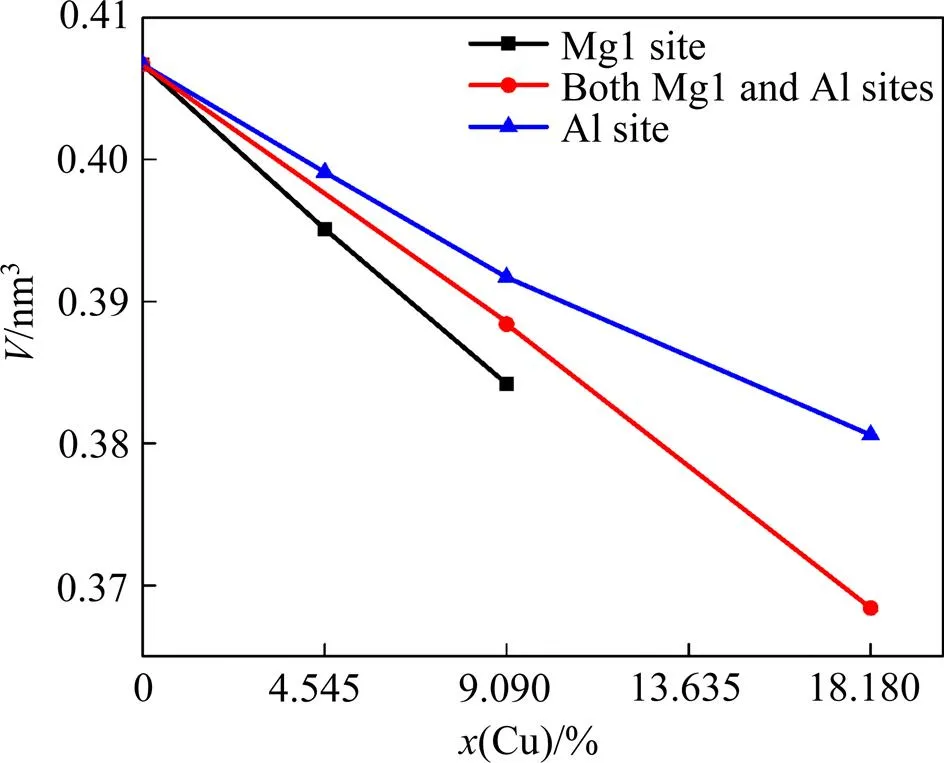
图2 掺Cu结构Mg5−xAl2−ySi4Cux+y的体积V与掺杂浓度、掺杂位置的关系
而本工作计算的Mg5Al2Si4的aʺ、bʺ和cʺ值分别为1.533 nm、0.408 nm及0.677 nm。与其相匹配的Al基体的各个方向的Al、Al、Al值分别为1.453 nm、0.405 nm和0.639 nm。所以,Mg5Al2Si4与Al基体的错配度δ、δ、δ分别为+5.5%、+0.7%和+5.9%。Cu掺入Mg5Al2Si4中改变了Mg5Al2Si4的晶胞参数和体积,所以晶格错配度也会变化。以掺杂浓度为4.545%的Mg5Al1.5Si4Cu0.5为例,计算其与Al基体的错配度,Mg5Al1.5Si4Cu0.5的、、值分别为1.522 nm、0.401 nm、0.681 nm,则δ、δ、δ分别为+4.7%,−1.0%和6.6%。比较Mg5Al2Si4、Mg5Al1.5Si4Cu0.5与Al基体的错配度,发现掺Cu后3个方向的错配度均有变化,且轴方向错配度都很小,、轴方向的错配度较高,其他掺Cu浓度的结构与Al基体的错配度也有此特点。这说明Cu掺杂前后ʺ与Al基体在轴方向匹配程度较高,在轴方向和轴方向与Al基体的匹配较差。而实验研究表明:ʺ与Al基体在轴方向完全共格,这表明该计算结果是准确的。
2.2 Cu的掺杂位置及掺杂浓度对掺Cu相稳定性的影响
形成焓常用来衡量相结构的稳定性。形成焓为负值,则相结构稳定;形成焓越负,对应的相结构越稳定,在合金中越容易形成。本文采用的形成焓计算公式[21]如下:
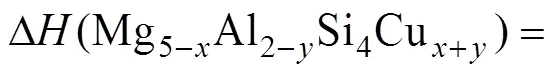
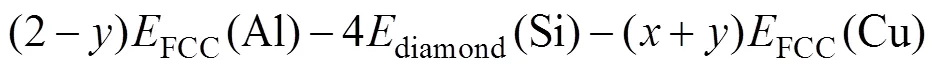
式(1)是计算Cu掺入Mg5Al2Si4后形成的Mg5−xAl2−ySi4Cu+y结构的形成焓公式。式中,total(Mg5−xAl2−ySi4Cu+y)代表Mg5−xAl2−ySi4Cu+y结构的总能量,HCP(Mg)、FCC(Al)、diamond(Si)和FCC(Cu)分别是几何优化后的单质块体材料总能量平均到每个原子后的能量。在本文计算中,Mg为HCP结构,Al、Cu为FCC结构,Si为金刚石立方结构。
不同掺杂浓度、掺杂位置的掺Cu相Mg5−xAl2−ySi4Cu+y的形成焓见图3。由图3可知,无论结构中是否掺杂Cu,Mg5−xAl2−ySi4Cu+y的形成焓均为负值,表明这些结构都能够在合金中形成,只是形成难易不同,形成焓越低的越容易形成。
从图3可以看出,相比于Mg5Al2Si4的形成焓Δ0,Cu 掺入Mg5Al2Si4后形成的Mg5−xAl2−ySi4Cu+y结构的形成焓Δ有的升高有的降低。ΔMg1(Cu只替代Mg1原子)升高,ΔAl(Cu只替代Al原子)降低。另外, ΔMg1-Al(Cu既替代Mg1又替代Al原子)也降低,结构稳定性的顺序为:ΔAl<ΔMg1-Al<Δ0<ΔMg1。该计算结果表明Cu既替代Mg1又替代Al原子和Cu只替代Al原子的结构在合金中更容易形成,而Cu只替代Mg1原子的结构在合金中不容易形成。

图3 掺Cu结构Mg5−xAl2−ySi4Cux+y的形成焓ΔH与掺杂浓度、掺杂位置的关系
有研究[13, 15]报道,在扫描透射电子显微镜的高角环形暗场像中观察到Mg5Al2Si4相的Mg1原子列和Al原子列中含有Cu;而DING 等[14]在Mg5Al2Si4相的Al原子列中发现了Cu的存在,在Mg1原子列中未发现Cu。即实验观察到的Cu可以既替代Mg1又替代Al原子,也可以只替代Al原子,这恰好证明了上述计算结果的准确性。
值得注意的是,ΔMg1和ΔMg1-Al对掺杂浓度不敏感,只有ΔAl对掺杂浓度敏感。这一点可以从图3中形成焓随掺杂浓度的变化看出。对于Cu只替代Mg1原子和既替代Mg1又替代Al原子的结构,掺杂浓度不同,形成焓变化很小,在0.5 kJ/mol范围内;而对于Cu只替代Al原子的结构,掺杂浓度不同,形成焓的变化较大,超过了1 kJ/mol。这说明Cu只替代Al原子的结构稳定性受掺杂浓度的影响较大,其他两种情况下掺杂浓度对结构稳定性的影响很小。
2.3 掺Cu相的电子结构性质
2.3.1 Cu的掺杂位置对掺Cu结构稳定性的影响机理
上述结果表明,Cu只替代Al的结构稳定性最高,其次是Cu既替代Mg1又替代Al的结构,Mg5Al2Si4的稳定性再其次,Cu只替代Mg1的结构稳定性最低,这是从能量的角度分析得到的结论。材料的物理性质由其微观电子结构决定,所以不同结构的稳定性差异取决于电子结构上的不同。为探索Cu的掺杂位置对掺Cu结构Mg5−xAl2−ySi4Cu+y稳定性的影响机理,以掺杂9.090% Cu为例,计算并比较Mg5AlSi4Cu(Cu只替代Al原子)、Mg4.5Al1.5Si4Cu(Cu既替代Mg1又替代Al原子)和Mg4Al2Si4Cu(Cu只替代Mg1原子)的总态密度,Mg5Al2Si4的总态密度图作为参考,如图4所示。由图4可知:1) 相比于Mg5Al2Si4的总态密度,掺Cu结构的总态密度图都在−5~0 eV范围内存在一个特别尖锐的峰,这是由Cu的局域化d电子引起的;2) 无论结构中是否掺入Cu,在费米能级处均存在赝能 隙[22]。且Mg5AlSi4Cu、Mg4.5Al1.5Si4Cu、Mg5Al2Si4和Mg4Al2Si4Cu 的赝能隙分别为2.483、2.376、2.272和2.079 eV。
由于赝能隙越宽,共价键越强,体系越稳定[23−24]。所以,Cu只替代Al的结构稳定性最高,其次是Cu既替代Mg1又替代Al的结构,Mg5Al2Si4的稳定性再其次,Cu只替代Mg1的结构稳定性最低。
2.3.2 掺Cu结构的成键特点
差分电荷密度图可以反映原子间成键方式。为研究Cu进入Mg5Al2Si4后原子间的成键变化,计算了未掺Cu的Mg5Al2Si4和9.090% Cu掺入Mg5Al2Si4后结构(Mg4Al2Si4Cu、Mg4.5Al1.5Si4Cu、Mg5AlSi4Cu)的差分电荷密度图,如图5所示。电荷密度采用蓝、绿、红标示,红色表示得电子区域,蓝色为失电子区域,绿色为中间态。由图5可知:1) 对于Mg5Al2Si4结构(图5(a)),两个Si1原子之间、Si1原子和Al原子之间、Si1原子和Mg1原子之间存在强烈的共价键,Si2原子和周围的Al原子、Mg1原子、Mg2原子之间存在较弱的共价键;原子之间的空隙处存在自由电子气,表明了金属键的存在;2) 掺Cu结构的差分电荷密度(见图5(b)~5(d))与未掺Cu结构的差分电荷密度图5(a)相比,除了被替代的Al原子或Mg1原子周围的电荷密度有变化,其他原子周围的电荷密度变化不大,所以结构中仍然存在共价键和金属键。所以Cu掺入Mg5Al2Si4后不改变原子间的成键方式,掺Cu前后结构中均有共价键和金属键。
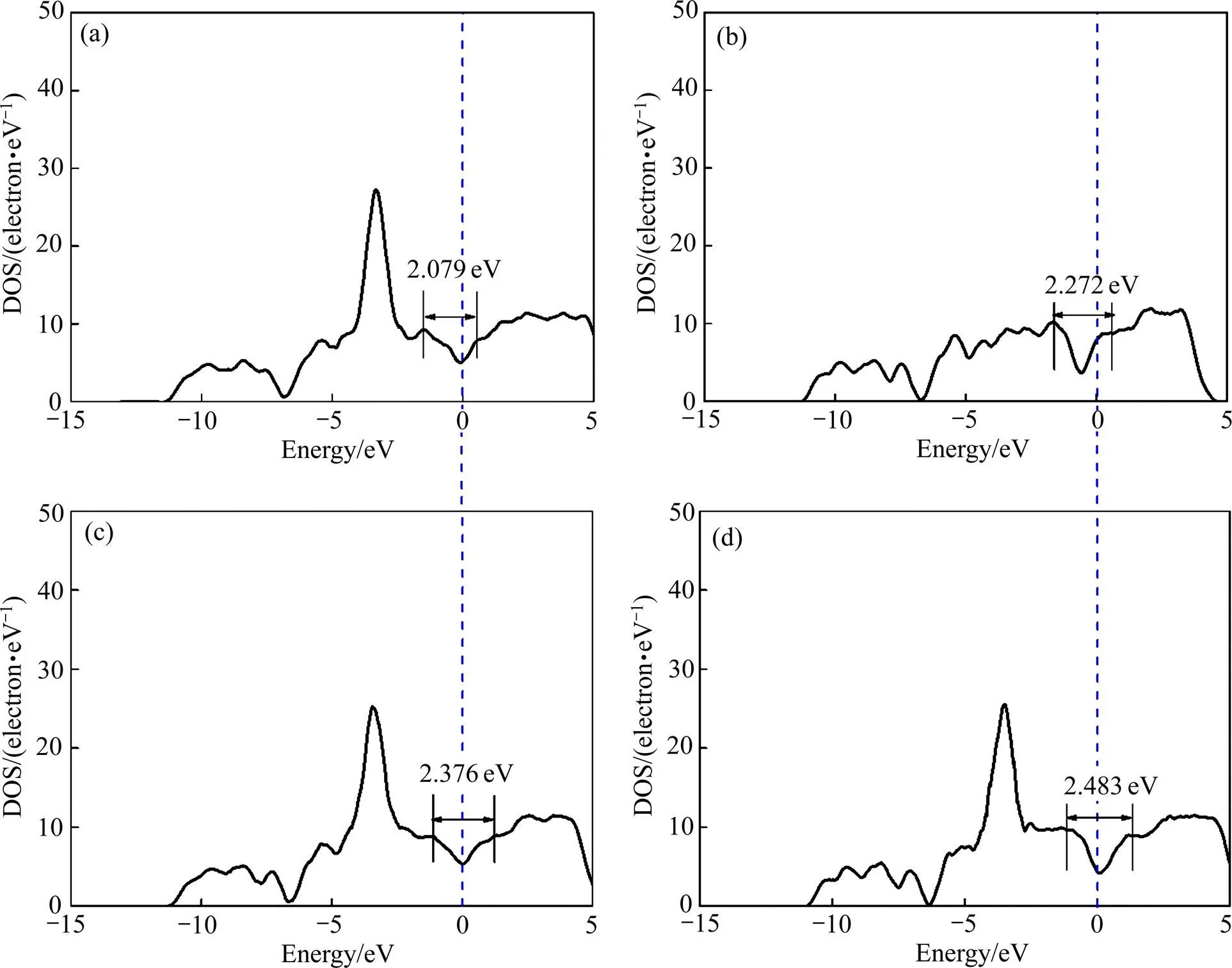
图4 Mg4Al2Si4Cu、Mg5Al2Si4、Mg4.5Al1.5Si4Cu和Mg5AlSi4Cu的总态密度
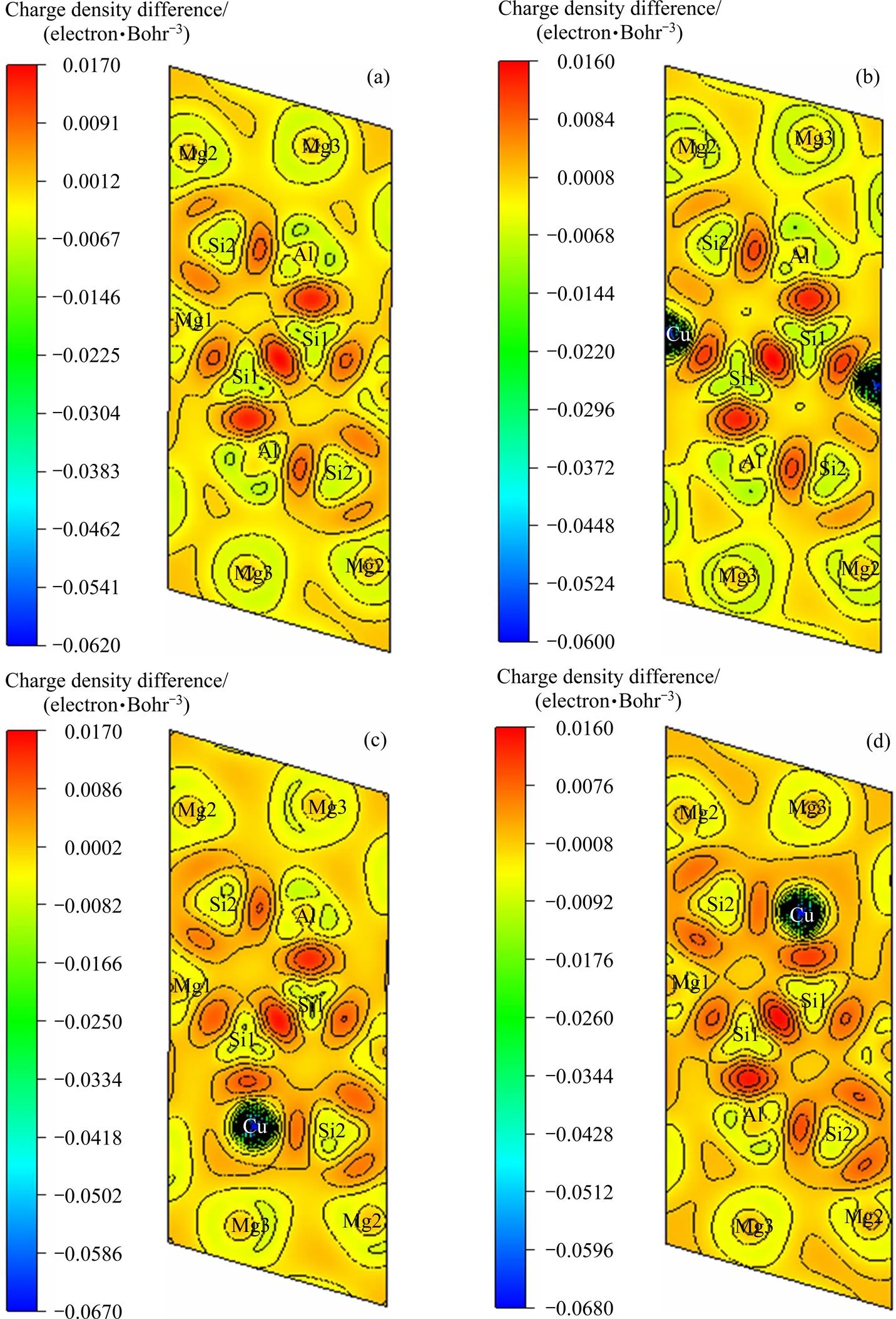
图5 不同相(010)面内的差分电荷密度图
3 结论
1) 未掺杂Cu的ʺ相的晶胞参数与文献报道相符。掺杂Cu后体系的晶胞形状发生微小变形且体积减小,并且掺杂浓度越大,体积越小。在相同掺杂浓度下,Cu掺杂位置不同的结构体积不同,其中Cu只替代Al原子的结构体积最大。体积差异归咎于替代原子Cu和被替代原子Mg1和Al的半径大小不同。Cu掺杂改变了ʺ相与Al基体的晶格错配度,其中ʺ相与Al基体在轴方向的错配度最小,说明ʺ相与Al基体在轴方向完全共格。
2) Cu既替代Mg1又替代Al原子和Cu只替代Al原子的结构在合金中更容易形成,而Cu只替代Mg1原子的结构在合金中不容易形成,该计算结果与实验报道相符。Cu只替代Al原子的结构稳定性受掺杂浓度的影响较大,而Cu只替代Mg1原子与Cu既替代Mg1又替代Al原子的结构稳定性几乎不受掺杂浓度的影响。
3) Cu掺入Mg5Al2Si4后不改变原子间的成键方式,掺杂Cu前后的结构中均有共价键和金属键。掺杂Cu后形成的Mg5−xAl2−ySi4Cu+y相结构的稳定性和体系在费米能级附近的赝能隙密切相关。
[1] HIRSCH J. Recent development in aluminum for automotive applications[J]. Transactions of Nonferrous Metals Society of China, 2014, 24(7): 1995−2002.
[2] CHEN J H, COSTAN E, van HUIS M A, XU Q, ZANDBERGEN H W. Atomic pillar based nanoprecipitates strengthen AlMgSi alloys[J]. Science, 2006, 312: 416−419.
[3] HASTING H K, LEFEBVRE W, MARIOARA C, WALMSLEY J C, ANDERSEN S J, HOLMESTAD R, DANOIX F. Comparative study of the″-phase in a 6xxx Al alloy by 3DAP and HRTEM[J]. Surface and Interface Analysis, 2007, 39: 189−194.
[4] CHEN Rui, XU Qing-yan, GUO Hui-ting, XIA Zhi-yuan, WU Qin-fang, LIU Bai-cheng. Modeling the precipitation kinetics and tensile properties in Al-7Si-Mg cast aluminum alloys[J]. Materials Science and Engineering A, 2017, 685: 403−416.
[5] WANG Zhi-xiu, LI Hai, MIAO Fen-fen, SUN Wang-jie, FANG Bi-jun, SONG Ren-guo, ZHENG Zi-qiao. Improving the intergranular corrosion resistance of Al-Mg-Si-Cu alloys without strength loss by a two-step aging treatment[J]. Materials Science and Engineering A, 2013, 590: 267−273.
[6] JAAFAR A, RAHMAT A, HUSSAIN Z, ZAINOL I. Effect of Mg, Si and Cu content on the microstructure of dilute 6000 series aluminum alloys[J]. Journal of Alloys and Compounds, 2011, 509: 8632−8640.
[7] MURAYAMA M, HONO K, MIAO W F, LAUGHLIN D E. The effect of Cu additions on the precipitation kinetics in an Al-Mg-Si alloy with excess Si[J]. Materials Science and Engineering A, 2001, 32: 239−246.
[8] QIU Yue, KONG Yi, XIAO Shi-di, DU Yong. Mechanical properties of″ precipitates containing Al and/or Cu in age hardening Al alloys[J]. Journal of Materials Research, 2016, 31: 580−588.
[9] ANDERSEN S J, ZANDBERGEN H W, Jansen J. Structure determination of Mg5Si6particles in Al by dynamic electron diffraction studies[J]. Science, 1997, 277: 1221−1225.
[10] HASTING H S, FRØSETH A G, ANDERSEN S J, VISSERS R, WALMSLEY J C, MARIOARA C D, DANOIX F, LEFEBVRE W, HOLMESTAD R. Composition of″ precipitates in Al-Mg-Si alloys by atom probe tomography and first principles calculations[J]. Journal of Applied Physics, 2009, 106(123527): 1−9.
[11] WENNER S, HOLMESTAD R. Accurately measured precipitate-matrix misfit in an Al-Mg-Si alloy by electron microscopy[J]. Scripta Materialia, 2016, 118: 5−8.
[12] FLAMENT C, RIBIS J, GARNIER J, SERRUYS Y, LEPRÊTRE F, GENTILS A, BAUMIER C, DESCOINS M, MANGELINCK D, LOPEZ A, COLAS K, BUCHANAN K, DONNADIEU P, DESCHAMPS A. Stability of″ nano-phases in Al-Mg-Si(-Cu) alloy under high dose ion irradiation[J]. Acta Materialia, 2017, 128: 64−76.
[13] BUCHANAN K, COLAS K, RIBIS J, LOPEZ A, GARNIER J. Analysis of the metastable precipitates in peak-hardness aged Al-Mg-Si(-Cu) alloys with differing Si contents[J]. Acta Materialia, 2017, 132: 209−221.
[14] DING Li-peng, JIA Zhi-hong, NIE Jian-feng, WENG Yao-yao, CAO Ling-fei, CHEN Hou-wen, WU Xiao-zhi, LIU Qing. The structural and compositional evolution of precipitates in Al-Mg-Si-Cu alloy[J]. Acta Materialia, 2018, 145: 437−450.
[15] LI Kai, BÉCHÉ A, SONG Min, SHA Gang, LU Xing-xu, ZHANG Kai, DU Yong, RINGER S P, SCHRYVERS D. Atomistic structure of Cu-containing″ precipitates in an Al-Mg-Si-Cu alloy[J]. Scripta Materialia, 2013, 75: 86−89.
[16] PERDEW J P, BURKE K, ERNZERHOF M. Generalized gradient approximation made simple[J]. Physical Review Letters, 1996, 77(18): 3865−3868.
[17] BLÖCH P E. Projector augmented-wave method[J]. Physical Review B, 1994, 50(24): 17953−17979.
[18] KRESSE G, JOUBERT D. From ultrasoft pseudopotentials to the projector augmented-wave method[J]. Physical Review B, 1999, 59(3): 1758−1775.
[19] ANDERSEN S J, ZANDBERGEN H W, JANSEN J, TRAEHOLT C, TUNDAL U, REISO O. The crystal structure of the″ phase in Al-Mg-Si alloys[J]. Acta Materialia, 1998, 46(9): 3283−3298.
[20] MATSUDA K, NAOI T, FUJII K, UETANI Y, SATO T, KAMIO A, IKENO S. Crystal structure of the″ phase in an Al-1.0mass%Mg2Si-0.4mass%Si alloy[J]. Materials Science and Engineering A, 1999, 262: 232−237.
[21] HU Hai, ZHAO Ming-qi, WU Xiao-zhi, JIA Zhi-hong, WANG Rui, LI Wei-guo, LIU Qing. The structural stability, mechanical properties and stacking fault energy of Al3Zr precipitates in Al-Cu-Zr alloys: HRTEM observations and first-principles calculations[J]. Journal of Alloys and Compounds, 2016, 681: 96−108.
[22] 孙顺平, 李小平, 雷卫宁, 王洪金, 汪贤才, 江海锋, 李仁兴, 江 勇, 易丹青. 基于第一性原理L12-Al3Sc点缺陷结构及成键行为的计算[J]. 中国有色金属学报, 2013, 23(8): 2147−2155. SUN Shun-ping, LI Xiao-ping, LEI Wei-ning, WANG Hong-jin, WANG Xian-cai, JIANG Hai-feng, LI Ren-xing, JIANG Yong, YI Dan-qing. Calculation of point defect structures and bonding behavior of L12-Al3Sc intermetallic based on first-principles[J]. The Chinese Journal of Nonferrous Metals, 2013, 23(8): 2147−2155.
[23] 傅 利, 赵宇宏, 杨晓敏, 王 楠, 文志勤, 韩培德. Mg-Al-Si-Ca合金系金属间化合物的电子结构和力学性能的第一性原理计算[J]. 稀有金属材料与工程, 2014, 43(11): 2733−2738. FU Li, ZHAO Yu-hong, YANG Xiao-min, WANG Nan, WEN Zhi-qin, HAN Pei-de. First principle calculation for electronic structure and mechanical properties of intermetallics in Mg-Al-Si-Ca alloy[J]. Rare Metal Materials and Engineering, 2014, 43(11): 2733−2738.
[24] 李燕峰, 徐 慧, 张 彪, 章立刚. Al-Sc金属间化合物的电子结构及稳定性和热力学性质[J]. 中国有色金属学报, 2010, 20(5): 946−953. LI Yan-feng, XU Hui, ZHANG Biao, ZHANG Li-gang. Electronic structure, stability and thermodynamic properties of Al-Sc intermetallics compounds[J]. The Chinese Journal of Nonferrous Metals, 2010, 20(5): 946−953.
First-principles study ofʺ phase in Cu doped 6000-series aluminum alloys
WEN Bo-yang1, JIA Zhi-hong1, 2, WU Xiao-zhi3, LIU Qing1
(1. College of Materials Science and Engineering, Chongqing University, Chongqing 400044, China; 2. Electron Microscopy Center, Chongqing University, Chongqing 400044, China; 3. College of Physics, Chongqing University, Chongqing 400044, China)
ʺ(Mg5Al2Si4) phase is the main strengthening phase in 6000-series aluminum alloys. The effect of Cu-doping on the geometrical structure, the phase stability and the electronic properties ofʺ(Mg5Al2Si4) phase were investigated by using projector augmented wave method and the generalized gradient approximation based on density functional theory. The results show that the calculated equilibrium lattice parameters ofʺ phase are in good agreement with available experimental results. After Cu incorporates intoʺ phase, the cell shape changes slightly and the cell volume decreases. Various doping contents and doping sites result in various geometrical properties of Cu-doped structures, which then impacts the lattice mismatch betweenʺ and Al matrix. The structures in which Cu atoms replace both Mg1 and Al atoms, or only replace Al atoms are easier to form in the alloys, while it is harder to form in the alloys for the structures, in which Cu atoms only replace Mg1 atoms, which is supported by experimental results. The analysis of electronic structures shows that the phase stability of the Cu-doped structures Mg5−xAl2−ySi4Cu+yis closely related to the pseudo gap near the Fermi level.
first-principles; 6000-series aluminum alloys; strengtheningʺ phase; phase structure stability; Cu-doping
Project(cstc2017zdcy-zdzxX0006) supported by the Special Major R&D Projects for Key Technology Innovation of Key Industries in Chongqing, China; Project(106112017CDJQJ308822) supported by Fundamental Research Funds for the Central Universities of China; Project(51421001) supported by Foundation for Innovative Research Groups of the National Natural Science Foundation of China
2018-02-28;
2018-07-02
JIA Zhi-hong; Tel: +86-23-65102029; E-mail: zhihongjia@cqu.edu.cn
重庆市重点产业共性关键技术创新专项重大主题专项项目(cstc2017zdcy-zdzxX0006);中央高校基本科研业务费项目(106112017CDJQJ308822);国家自然科学基金创新群体项目(51421001)
2018-02-28;
2018-07-02
贾志宏,教授,博士;电话:023-65102029;E-mail: zhihongjia@cqu.edu.cn
10.19476/j.ysxb.1004.0609.2018.10.05
1004-0609(2018)-10-1991-08
TG146.2
A
(编辑 李艳红)
