双唑草酮原药高效液相色谱分析方法研究
2018-11-16姜宜飞
傅 黎,黄 伟,姜宜飞*
(1.湖南加法检测有限公司,湖南 长沙 410014;2.农业部农药检定所,北京 100125)
1 前言
双唑草酮,CAS号1622908-18-2,分子式C20H19F3N4O5S。双唑草酮是一种新型HPPD类除草剂,与当前麦田常用的双氟磺草胺、苯磺隆、苄嘧磺隆、噻吩磺隆等ALS抑制剂类除草剂,唑草酮、乙羧氟草醚等PPO抑制剂类除草剂以及2甲4氯钠、2,4-D等激素类除草剂不存在交互抗性,可以有效解决抗性以及多抗性的播娘蒿、荠菜、野油菜、繁缕、牛繁缕、麦家公等阔叶杂草。
目前国内有关双唑草酮的分析方法尚未见任何公开报道。本文采用高效液相色谱法,对双唑草酮原药进行定量分析,该方法操作简便、快速、准确,分离效果好,准确度和精密度均能达到定量分析的要求。
2 试验部分
2.1 试剂和溶液 乙腈(色谱纯);磷酸(分析纯);超纯水(电阻率18.2MΩ·cm,25℃);双唑草酮标样,已知质量分数98.2%(由农业部农药检定所提供);双唑草酮原药(由某公司提供)。
2.2 仪器 高效液相色谱仪:Agilent 1100,具有二极管阵列检测器和自动进样器;Agilent色谱工作站;Millipore超纯水制备系统;色谱柱:250mm×4.6mm(id)不锈钢柱,内装ZORBAX SB-C185.0μm填充物。
2.3 液相色谱操作条件 流动相:ψ(乙腈∶0.1%磷酸水)=60∶40;流量∶1.0mL/min;柱温:30℃;检测波长:254nm;进样体积:5μL;保留时间:双唑草酮约4.0min。
双唑草酮原药的高效液相色谱图(图1)。
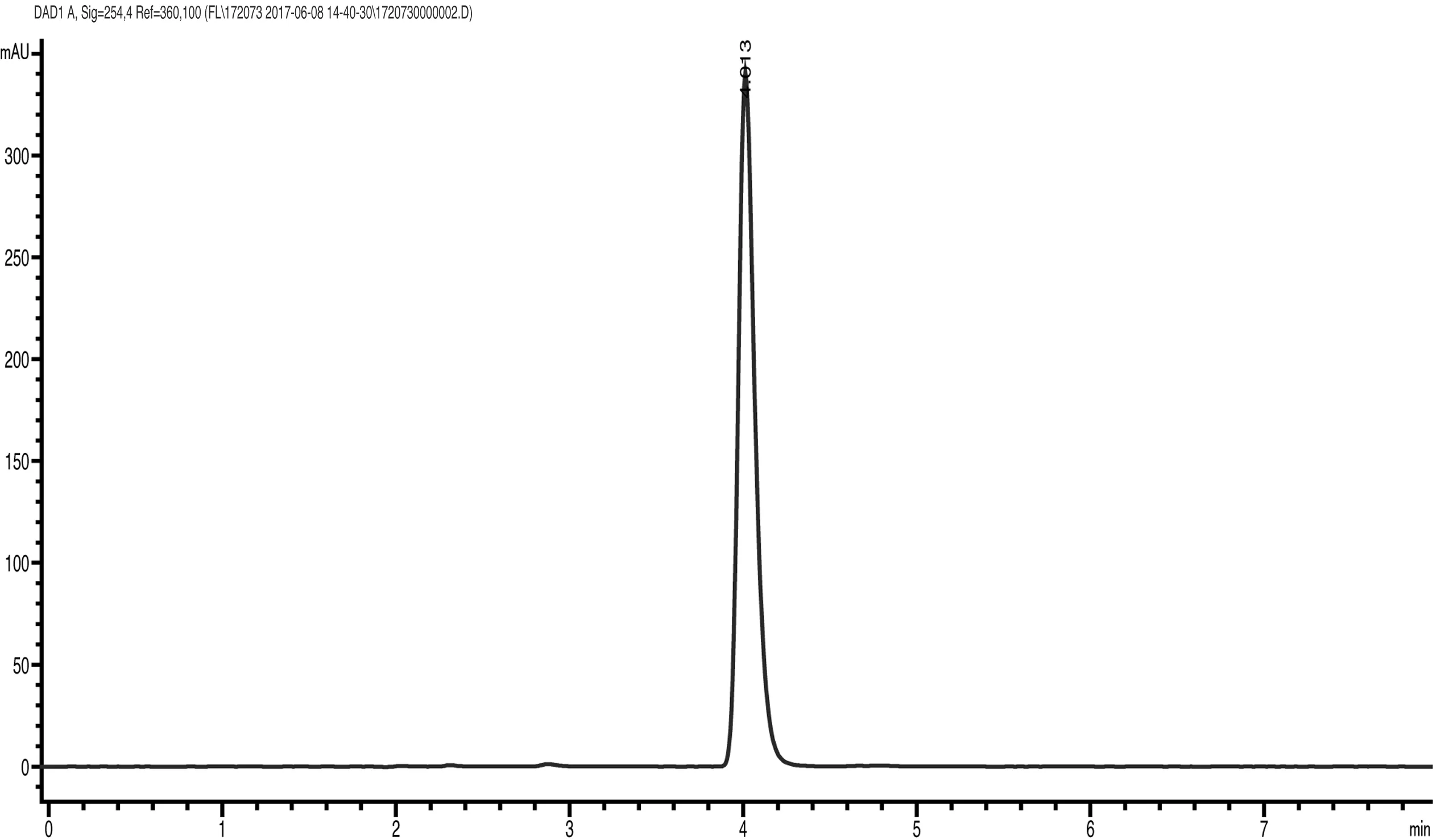
图1 双唑草酮原药高效液相色谱图
2.4 测定步骤
2.4.1 标样溶液的配制 称取双唑草酮标样0.02g(精确至0.000 2g),置于100mL容量瓶中,用乙腈溶解并稀释至刻度,摇匀。
2.4.2 试样溶液的配制 称取含双唑草酮0.02g的试样(精确至0.000 2g),置于100mL容量瓶中,用乙腈溶解并稀释至刻度,摇匀。
2.4.3 测定 在上述操作条件下,待仪器基线稳定后,连续注入数针标样溶液,直至相邻2针标样溶液的响应值相对变化<1.5%后,按照标样溶液、试样溶液、试样溶液、标样溶液的顺序进行测定。
2.4.4 计算 将测得的2针试样溶液以及试样前后2针标样溶液中双唑草酮峰面积分别进行平均。试样中双唑草酮的质量分数ω(%),按式(1)计算:
(1)
式中:
A1──标样溶液中双唑草酮峰面积的平均值;
A2──试样溶液中双唑草酮峰面积的平均值;
m1──标样的质量,g;
m2──试样的质量,g;
P──标样中双唑草酮的质量分数,%。
3 结果与讨论
3.1 色谱条件的选择 通过Agilent 1100高效液相色谱仪的光谱数据采集功能,获得双唑草酮的紫外波长扫描图(图2)。从图中可以看到双唑草酮最大吸收波长在210nm处,在254nm处也有较大的吸收,为了尽量减少溶剂的干扰,将检测波长定为254nm。

图2 双唑草酮紫外吸收谱图
色谱柱选择常规的ZORBAX SB-C18反相柱。依据双唑草酮物化性质,用乙腈作为溶剂溶解样品,并选择乙腈和磷酸溶液作为流动相。将流动相按不同比例在色谱柱上进行试验,最终确定流动相为ψ(乙腈∶0.1%磷酸水)=60∶40,在流速1.0mL/min时,有效成分与杂质能得到很好的分离,峰形对称,基线平稳,能在短时间能得到满意的分析结果,提高了工作效率。
3.2 分析方法的线性相关性试验 将2.4.1中配制的标样溶液在上述色谱操作条件下进行分析,进样量分别为1、2、5、10、20μL,以双唑草酮的质量为横坐标,峰面积为纵坐标,绘制校正曲线。试验测得双唑草酮线性方程为y=20 150.521 2 x+3.526 4,相关系数为1.000 0。结果表明双唑草酮在测试的质量浓度范围内线性关系良好。
3.3 分析方法的精密度试验 从同一产品中准确称取5个试样,在上述色谱操作条件下进行分析,测得双唑草酮的标准偏差为0.16,变异系数为0.17%(表1)。

表1 分析方法的精密度试验结果
3.4 分析方法的准确度试验 从已知质量分数的双唑草酮(96.95%)中称取5个试样,分别加入一定量的双唑草酮标样(98.20%),在上述色谱操作条件下进行分析,测得双唑草酮的平均回收率为99.75%(表2)。

表2 分析方法的准确度试验结果
4 结论
试验建立了高效液相色谱法检测双唑草酮中有效成分的分析方法。试验结果表明,双唑草酮在测试浓度范围内线性关系良好,方法准确度和精密度较高,具有操作简便、快速的特点,是产品质量控制和应用研究中较为理想的分析方法。
