内质网应激和自噬的交互作用及其在阿尔茨海默病进展与防治中的作用
2018-11-12黄倩倩温彬宇赵永烈
黄倩倩,温彬宇,闫 妍,赵永烈,马 涛
(1. 北京中医药大学第三附属医院脑病科,北京 100029;2. 北京中医药大学东方医院实验中心,北京 100078)
阿尔茨海默病(Alzheimer’s disease, AD)起病隐匿,是一种以进行性记忆力丧失、认知功能障碍、人格和行为异常等为主要临床表现的神经退行性疾病,主要病理改变包括以β淀粉样蛋白(amyloid β, Aβ)沉积为主要成分的老年斑(senile plaque, SPs)和异常磷酸化Tau蛋白聚集形成的神经纤维缠结(neurofibrillary tangles, NFTs)[1]。内质网应激(endoplasmic reticulum stress, ERS)是细胞针对外界损伤的一种保护性机制,在AD病程中,Aβ和NFTs的过度累积均可激活ERS,引发未折叠蛋白反应(unfolded protein response, UPR),进而激活自噬,清除错误折叠蛋白[2]。大量研究显示,在AD中,由于ERS的持续激活及自噬功能障碍,导致过量累积的Aβ与NFTs不能被有效清除,从而造成神经元损伤,导致认知记忆功能障碍[3]。近年研究发现,ERS与自噬的交互作用在AD发病及治疗中发挥关键作用,成为AD治疗的重要潜在靶标,受到日益关注。本文主要针对近年来ERS与自噬交互作用在AD发病及防治中的作用机制及研究进展做一综述。
1 ERS与AD
内质网是真核细胞中负责膜蛋白合成、初始翻译后修饰、折叠和分泌的细胞器。当未折叠或错误折叠蛋白在内质网腔中积累,内质网蛋白折叠功能过载时,会造成内质网稳态失衡,诱发ERS,进而激活UPR,引发下游一系列反应[4]。UPR分别由3个跨膜感受蛋白介导的通路启动,这3个蛋白分别是蛋白激酶样内质网激酶[protein kinase RNA (PKR)-like ER kinase, PERK]、肌醇需求酶1α(inositol-requiring enzyme 1α, IRE1α)和活化转录因子6(activating transcription factor 6, ATF6)。在正常生理状态下,这3种蛋白均与葡萄糖调节蛋白78(glucose-regulated protein 78, GRP78)结合,保持无活性状态。ERS发生时,PERK、IRE1α、ATF6与GRP78解离并被激活,启动UPR,上调包括分子伴侣在内的UPR相关基因表达,下调总体蛋白质翻译水平,以恢复内质网内环境稳态,保护细胞功能[5]。
大量研究证明,ERS是AD的核心病变。在AD患者大脑中,ERS标志物GRP78水平明显增高,并且与AD患者的Braak分期呈正相关;UPR中关键蛋白,包括磷酸化的PERK、真核翻译启始因子2α(eucaryotic translation initiation factor 2α, eIF2α)、IRE1α等水平,在AD患者脑中均明显增加。研究表明,UPR发生在AD早期阶段,随着病情加重,内质网稳态持续失衡,适应性、保护性ERS逐渐转变为持续性、损伤性ERS,造成神经元凋亡和突触缺失,损害认知和学习记忆能力[3]。Marcora等[6]发现,伴随着AD模型果蝇的衰老,UPR的保护作用减弱,而PERK持续磷酸化导致的神经变性损伤明显增强,ERS由适应性、保护性向持续性、损伤性转变。AD患者及模型小鼠脑中,eIF2α持续磷酸化导致的持续性ERS,可抑制突触蛋白的合成,降低神经突触可塑性,加重记忆功能障碍[7]。
Aβ的毒性积聚是AD病变的重要因素,Aβ的过量累积可导致SPs形成,造成突触缺失、神经功能失调和神经元死亡。内质网内环境与Aβ产生和加工密切相关,Aβ是淀粉样前体蛋白(amyloid precursor protein, APP)通过β-分泌酶1(beta-secretase 1, BACE1)和γ-分泌酶依次剪切形成的短肽。正常细胞中,APP与GRP78结合,从而抑制Aβ产生;而在AD病理状态下,由于ERS失衡,APP转运异常,造成APP的异常积聚。Liu等[8]发现,衣霉素诱导的ERS使AD模型细胞UPR被激活,APP水平下降;但当ERS平衡破坏后,BACE1和γ-分泌酶复合物的核心组分-早老素1(presenilin 1,PS1)的表达会随之上调,致使Aβ生成增加,并进一步累积。在AD模型小鼠中,随着eIF2α磷酸化水平的增高,活化转录因子4(activating transcription factor 4, ATF4)和BACE1的表达水平也随之上升,Aβ生成和SPs沉积明显增加[9]。
PS1基因突变是AD的重要遗传致病因素,可以对ERS产生多种影响。PS1突变可以使GRP78的水平降低,抑制IRE1α、PERK的自身磷酸化和减弱ATF6信号通路,进而削弱UPR保护作用,导致ERS失衡,增加神经元对ERS损伤的易感性,进一步加重AD病理损伤[3]。Genereux等[10]发现在AD中,ERS激活ATF6信号通路后,PS1突变使分子伴侣ERdj3(ER-localized DnaJ homolog 3)的产生减少,导致Aβ分泌到胞外,并形成毒性聚集,破坏神经元功能,进而削弱UPR对内质网稳态的再平衡作用。
2 自噬与AD
细胞自噬是维持细胞内稳态,降解错误折叠蛋白和受损细胞器的重要代谢过程,是实现细胞自我更新的一种重要机制[3]。自噬根据降解途径的不同分为3类,即分子伴侣介导的自噬 (chaperon-mediated autophagy,CMA)、微自噬(microautophagy)和巨自噬(macroautophagy)。巨自噬主要通过自噬溶酶体来清除错误折叠蛋白质,以维持细胞稳态,本文所涉及的自噬主要是巨自噬。巨自噬对于神经元维持内稳态发挥着至关重要的作用,影响其功能的关键步骤主要是自噬体在神经元内的运输,以及自噬体与溶酶体的融合。自噬过程受到多种信号通路的调控。
自噬功能障碍与AD发病过程密切相关,自噬是清除异常错误蛋白聚集体的重要途径,健康神经元中的自噬囊泡通过与溶酶体结合,降解错误蛋白;而在AD中,自噬功能出现障碍,自噬囊泡大量累积,这可能是Aβ和磷酸化Tau蛋白异常累积的重要原因[11]。自噬溶酶体系统是Aβ的主要降解途径,在正常情况下,Aβ被自噬囊泡包裹后,自噬囊泡沿轴突的微管系统逆向运输到溶酶体部位,与之融合并降解Aβ。研究发现,AD患者中枢神经元的部分轴突和树突部位,自噬囊泡由于运输障碍导致大量堆积,造成自噬功能损伤,使轴突和树突部位异常肿胀[15]。同时,溶酶体功能障碍也是导致AD自噬功能障碍的重要原因,会造成自噬系统降解Aβ能力下降,Aβ无法有效清除而积聚,从而加剧AD病理损伤。研究表明,在溶酶体特异性水解酶组织蛋白酶B(cathepsin B)基因敲除小鼠脑中,Aβ产生明显增多;在AD患者及模型动物脑中,溶酶体功能异常往往出现于Aβ形成和AD临床症状出现之前,提示溶酶体功能损伤导致的自噬障碍发生在AD早期[12]。
雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)、微管相关蛋白1轻链3蛋白[microtubule-associated protein 1 (MAP1) light chain 3, LC3]和自噬激活蛋白Beclin-1等参与自噬调节的关键蛋白,均与AD的发生存在密切关系。mTOR对自噬发挥负调控作用,抑制mTOR,可以启动细胞自噬反应。Aβ沉积可以诱导mTOR信号激活,从而抑制自噬反应。通过mTOR诱导的自噬可以清除Aβ毒性蛋白质聚集体,发挥神经保护作用,是AD防治的重要靶标。 Majumder等[13]发现,抑制mTOR可以激活自噬通路,使UNC-51样激酶1(UNC-51 like kinase 1, ULK1)激酶复合物去磷酸化后激活,招募磷脂酰肌醇-3-激酶(phosphatidyl inositol 3 kinase, PI3K)复合物到内质网。使自噬相关蛋白(autophagy-related, ATG)12与 ATG5蛋白偶联,进而使LC3-I蛋白与磷脂酰乙醇胺结合,形成LC3-II,形成自噬小体,清除Aβ蛋白沉积和Tau形成的NFTs,减轻认知功能损伤。Beclin-1是激活自噬过程的重要调控因子,在Beclin-1敲除小鼠受影响的大脑区域中,神经元的自噬调节被破坏,影响APP代谢,导致细胞外Aβ蛋白异常沉积,提示上调Beclin-1对防止AD早期病变发挥重要作用。
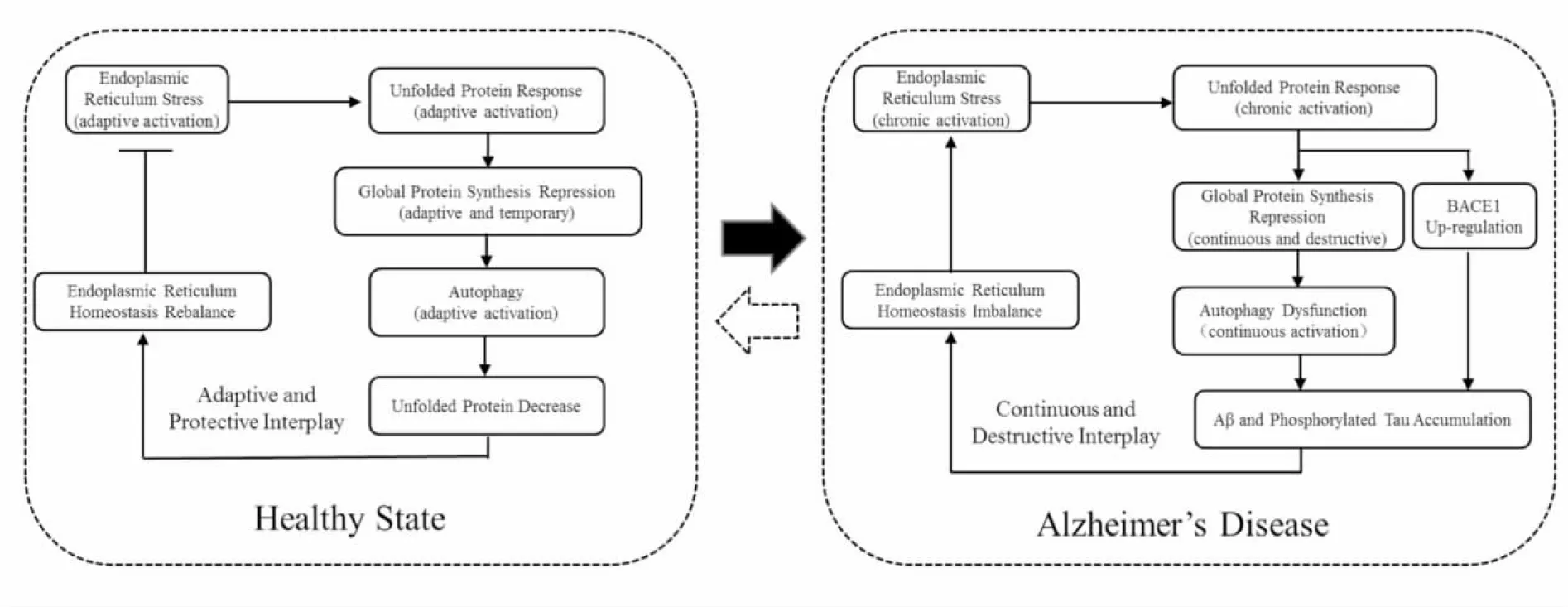
Fig 1 Crosstalk of ERS and autophagy in AD progress
3 内质网应激和自噬的交互作用与AD发生、发展的关系
ERS下的大量错误折叠蛋白累积引起的UPR可激活细胞自噬,而细胞自噬通过降解错误蛋白,恢复内质网稳态,反过来抑制ERS,减少细胞凋亡,促进细胞生存。由此,ERS与自噬的适度激活形成了机体内适应性、保护性的交互作用[14]。在AD发病过程中,这种适应性、保护性的交互作用逐渐转变为一种持续性、破坏性的交互作用(Fig 1)。其中,PERK、IRE1α及ATF6信号通路介导的ERS和自噬的交互效应,在AD发生、发展中发挥了重要的核心作用(Fig 2)。
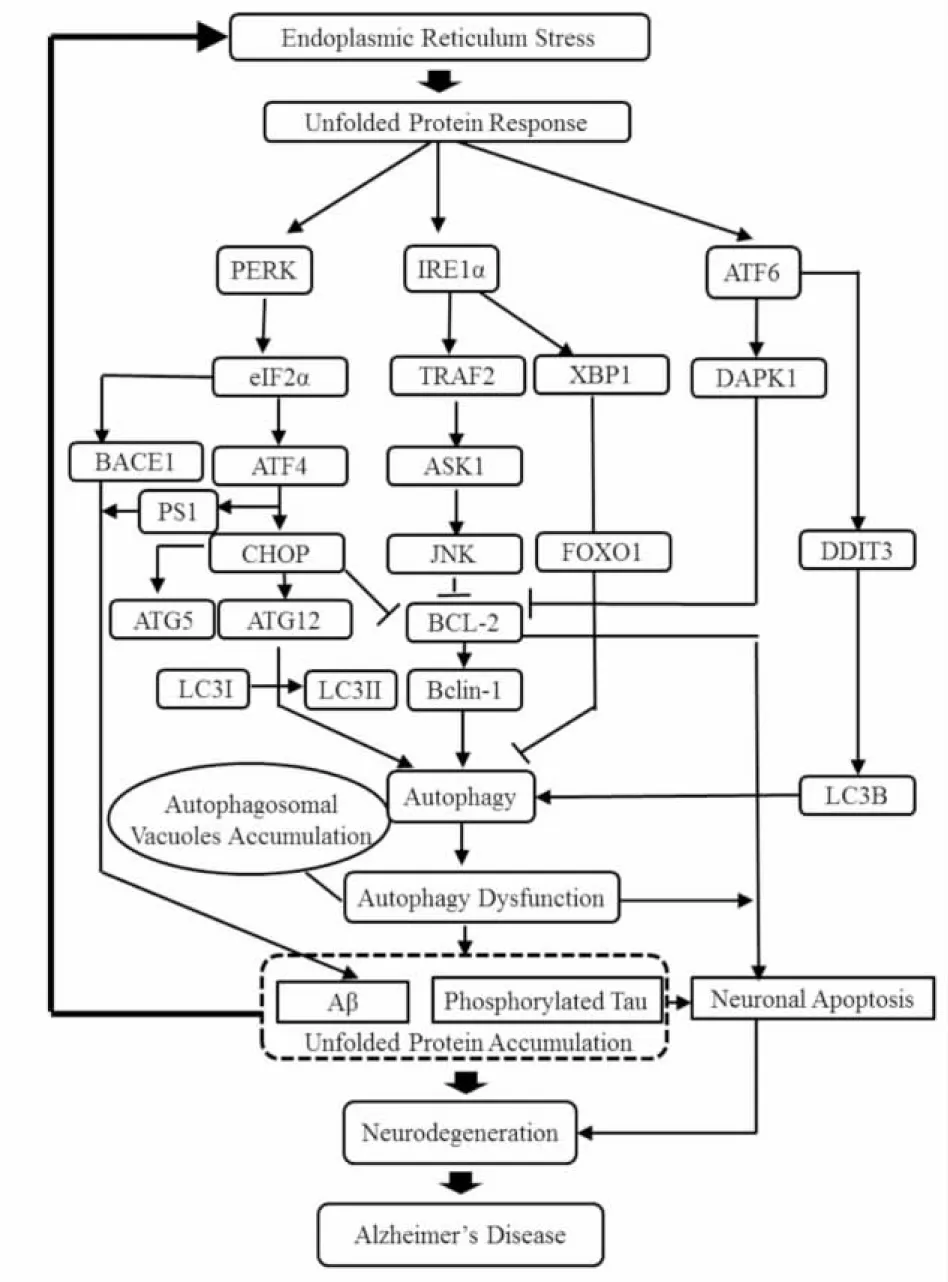
Fig 2 Pathways involved in crosstalk between ERS
3.1PERK/eIF2α介导的ERS和自噬交互作用许多研究证明,PERK/eIF2α途径是介导ERS与自噬交互作用的重要调控因子。Rouschop等[15]发现,PERK信号通路激活的ATF4和CCAAT/增强子结合蛋白同源蛋白(CCAAT/enhancer-binding protein-homologuous protein, CHOP),能够上调自噬相关蛋白LC3B和ATg5基因的转录,从而促进自噬体的形成。ERS还能够通过eIF2α-ATF4-CHOP途径,调控自噬蛋白ATG12,促使 LC3I转变为 LC3II,诱发细胞自噬。PERK是激活eIF2α的重要激酶,与细胞的自噬反应密切相关。在AD中,持续性ERS激活PERK-eIF2α信号通路,磷酸化eIF2α上的51位丝氨酸,下调总体蛋白质翻译水平;激活的eIF2α上调ATF4 mRNA,进而上调CHOP等转录因子,抑制抗凋亡蛋白B细胞淋巴瘤蛋白2(B-cell lymphoma 2, Bcl-2)基因转录,上调Beclin-1表达,从而导致自噬启动[9]。Urra等[16]发现,持续性ERS使CHOP持续上调,通过抑制Bcl-2和上调促凋亡蛋白Bcl-2相关蛋白x(Bcl-2-associated x, Bax)、Bcl-2同源拮抗蛋白(Bcl-2 homologous antagonist/killer, Bak),加速细胞凋亡,加重AD相关的神经变性和记忆障碍。持续性ERS导致eIF2α过度磷酸化,使细胞总体蛋白质合成下降,损害神经功能;同时上调BACE1,导致Aβ过量产生和SPs沉积[17]。在AD转基因小鼠中,持续性激活的PERK-eIF2a能够上调BACE1的表达,影响APP的加工过程,增加Aβ的沉积;累积的Aβ反过来进一步激活ERS,加重神经损伤[18]。AD中持续性自噬损伤导致Aβ降解能力下降,造成Aβ累积增加,反过来也会加重ERS。Ohta等[19]发现,Atg5基因敲除的HEK293细胞由于自噬功能障碍,会激活PERK/eIF2α途径,导致ATF4激活,上调PS1表达,提高γ-分泌酶活性,增加Aβ生成,造成ERS持续激活。
3.2IRE1α介导的ERS和细胞自噬交互作用IRE1α信号通路介导的ERS和自噬的交互作用,与AD的病理过程密切相关。IRE1α是一种具有核酸内切酶活性的I型跨膜蛋白,在通过自身二聚化和磷酸化激活后,IRE1α对无活性的X-盒结合蛋白1(X box binding protein-1, XBP1)mRNA进行加工,使其翻译成有活性的转录因子XBP1,可调节多个UPR靶基因的表达。另外,IRE1α信号通路激活的XBP1可增加ER中伴侣分子的表达,促进错误蛋白质的折叠和降解,以恢复内质网稳态。ERS激活的IRE1α信号通路与自噬的激活密切相关。IRE1α激活后,与肿瘤坏死因子受体相关因子2 (tumor necrosis factor receptor-associated factor 2, TRAF-2)结合,募集凋亡信号调节激酶1(apoptosis signal-regulating kinase 1, ASK1),随后激活c-Jun氨基末端激酶(c-Jun NH2-terminal kinase, JNK),进而磷酸化Bcl-2,激活Beclin-1,诱发自噬反应。在AD中,由于Bcl-2磷酸化失活,导致神经元凋亡,加重AD神经损伤。Vogel等[20]发现,ASK1介导的JNK激活后,还可调节APP的加工,增加细胞内Aβ的积累,导致神经损伤。但同时在ERS中,XBP1与转录因子FOXO1的结合,会对自噬起到负性调节作用。Safra等[21]研究发现,秀丽线虫AD模型中,敲除XBP1基因,可以上调自噬,减少Aβ毒性累积造成的损伤,表明XPB1是AD治疗的重要潜在靶标。另外,XBP1还可以与γ-分泌酶复合体的负性调控因子UBQLN1基因启动子结合,控制APP的分装与加工,影响Aβ的生成。
3.3ATF6介导的ERS和细胞自噬交互作用ATF6信号通路在ERS与自噬的交互作用中发挥着重要作用。ATF6是II型跨膜蛋白,属于亮氨酸拉链家族(bZIP)结构域中的转录因子,在内质网内,与GRP78结合而保持非活性状态。ERS发生后,去除 GRP78的ATF6被转运到高尔基体上,在高尔基体内,ATF6被特定的蛋白酶S1P(site 1 protease)和S2P(site 2 protease)切割后激活,之后移位到核内,参与调控多个UPR相关基因的表达。研究发现,ATF6激活后,可以上调UPR相关蛋白GRP78和葡萄糖调节蛋白94(glucose-regulated protein 94, GRP94)基因的表达,减弱神经元对ERS易感性,保护神经元,改善认知功能水平,显示ATF6可以做为AD潜在治疗靶标。有研究表明,在ERS导致的UPR中,ATF6上调死亡相关蛋白激酶1(death-associated protein kinase 1, DAPK1),激活的DAPK1磷酸化Beclin-1,促进其与Bcl-2解离,激活自噬,同时促进细胞凋亡[3]。ATF6的激活上调DNA损伤诱导转录因子3(DNA damage inducible transcript 3, DDIT3)的表达,DDIT3可与自噬相关蛋白LC3B基因上游-253-99启动子区结合,上调LC3B的表达,促进自噬。当AD发生时,持续性激活的ERS,导致ATF6信号通路受损,致使分子伴侣ERdj3的产生减少,导致Aβ生成增加,破坏神经元功能,加重AD病理损伤[10]。
4 结语与展望
综上所述,ERS与自噬广泛参与了AD的生理和病理过程,两者的交互作用与AD的发生、发展密切相关。ERS与自噬适应性、保护性的交互作用可以帮助细胞清除毒性蛋白聚集体,减轻内质网腔内失衡压力,恢复细胞稳态。在AD中,内质网稳态的持续失衡和自噬功能障碍,导致ERS与自噬之间的上述交互作用逐渐转变为一种持续性和破坏性的交互作用。AD中,自噬功能障碍导致Aβ及NFTs降解下降并异常累积,反过来进一步加重ERS的持续激活。首先,功能障碍的自噬长时间过度激活,导致自噬囊泡不断聚积,被损害的自噬溶酶体系统面临重大负荷,损伤神经元;其次,eIF2α的持续磷酸化,使蛋白总体合成减少,导致神经元正常生理活动受阻,损害神经系统功能,如eIF2α的持续磷酸化可使突触相关蛋白表达下降,破坏突触可塑性[7];再次,eIF2α的持续磷酸化导致BACE1上调,使Aβ产生增加和过量累积形成SPs,破坏神经功能;最后,ERS与自噬的恶性循环激活凋亡信号通路,诱导神经元凋亡,加重神经变性,推动AD病程恶化(Fig 1)。
由于ERS与自噬持续性、破坏性的交互作用在AD发生及进展中的核心作用,因此成为当前AD防治极有价值的潜在靶标,为AD治疗和干预开辟了新的思路。针对PERK/eIF2α通路调控,Halliday等[22]发现ATF4抑制剂ISRIB可以下调 ATF4表达,恢复细胞蛋白合成水平,明显提高大鼠的认知能力,且ISRIB能透过血脑屏障,表现出良好的生物利用度。小分子物质Salubrinal和Sephin 1通过阻止eIF2α磷酸化,抑制ERS,提高蛋白合成水平,恢复自噬功能,发挥神经保护作用[23]。研究发现,针对IRE1α核酸内切酶活性的抑制剂,如4μ8c、MKC-3946、SFT-083010、色霉素A3和ATP-竞争性抑制剂如APY29、sunitinib,以及天然活性物质如槲皮素、白藜芦醇等,均可以通过调控IRE1α通路,调节ERS,发挥治疗作用,体现出巨大的潜在临床应用价值[24]。直接调控ATF6表达或活性的化合物,目前还未见报道,但研究表明,Nucleobindin 1可以通过抑制SP1活性,阻止ATF6活化。大量抗氧化活性的天然产物,如黄酮类的堪非醇等,可以通过下调ATF6,保护机体免受ERS过度激活的损伤,具有很好的临床应用前景[25]。
综上所述,ERS与自噬的持续性、破坏性交互作用是AD发生、发展的重要因素,针对这一病理过程进行干预,对于缓解AD病程具有重要意义,因此成为治疗AD极具应用前景的药物靶标,为未来AD治疗药物的研发指明了重要方向。
