基于叶绿体DNA非编码序列的蒙古扁桃谱系地理学研究
2018-11-06段义忠白春梅段春燕申烨华
段义忠,白春梅,段春燕,申烨华*
(1 榆林学院 陕西省陕北矿区生态修复重点实验室,陕西榆林 719000; 2 西北大学 化学与材料学院,西安 710069)
第三纪以来古地中海的气候变迁以及青藏高原的快速隆升引起了古地中海植物区系乃至全球植物分布格局的变化[1]。对于当前物种而言,影响种群历史动态及空间分布格局比较大的地质历史事件主要是第四纪气候的波动[2-3]。在第四纪,欧洲、北美和北极地区的冰川发育比较突出,气候变化较大,从而影响了这些地区物种的分布。在植物类群的历史发展过程中,不同地区发生不均一的灭绝、进化以及迁移,导致了世界不同地区植物区系上的多样性和复杂性,因此不同植物区系中特有现象的明显程度、性质、起源、演变及组成有所不同[4],特别是末次盛冰期结束以来,气候的变化打乱了物种在时间尺度上的进化,改变了物种的空间分布格局[5]。谱系地理学(phylogeography)是由Avise等[6]首先提出,主要研究物种进化(强调近缘种或种内居群间的谱系)与地质历史事件(如高原、冰期、山岳隆升及地理隔离)的相互关系,该学科强调的是历史因素和基因分子系统对当前物种分布格局的影响,属于生物地理学(historical phylogeography)的一个分支[7-8]。与传统的生物地理学相比,谱系地理学不仅仅局限于解释现有居群的分布状况,而是更深入地探究居群形成现有分布格局的历史成因,阐述其进化过程,分析区域类群在时间和空间尺度上的动态变化,进而重建生物区系的历史[9]。尤其在第四纪,欧洲、北美和北极地区的冰川发育比较突出,冰期与间冰期气候反复波动,导致环境发生剧烈变化,从而影响全球特别是北半球动植物的群体遗传结构及地理分布格局[10-12]。
当前中国的植物谱系地理学研究主要集中在东亚以及北半球植物区系的起源、演化及其分子生物地理学,中国-喜马拉雅重要和群落特征成分的时空演化规律的研究上,尤其关于青藏高原及其毗邻地区的植物分子谱系地理学发展较为迅猛[13-14]。青藏高原与周边地区形成现有植物分布格局大致分为两种模式:一种模式为青藏高原东南部边缘是高山植物在第四纪冰期的避难所,高原台面的居群是间冰期或冰后期由东南部避难所扩散而来,代表物种为祁连圆柏(Juniperusprzewalskii)、青海云杉(Piceacrassifolia)、窄叶鲜卑花(Sibiraeaangustata)、山蓼(Oxyriadigyna)、蒙古绣线菊(Spiraeamongolica)等[15-19];另外一种模式为某些耐寒的植物第四纪冰期时在高原台面若干个相互隔离的避难所内保留下来,并且发生不同程度的异域分化,代表植物是露蕊乌头(Aconitumgymnandrum)、金露梅(Potentillafruticosa)及西川红景天(Rhodiolaalsia)等[20-22]。目前关于中国西北干旱区植物谱系地理学研究也有了不少积累[23-25],由于中国大部分地区处于中低纬度,在第三纪后期和整个第四纪没有被大面积的山地冰川覆盖,但持续的高度隆升所引起的剧烈气候动荡促进了中国西北部大部分地区的干旱化和沙漠扩张,很大程度上影响了该地区的水文和气候,也必将对该地区植物的地理分布格局和进化历史产生深刻影响[26-27]。孟宏龙等[28]对中亚-古地中海植物区系典型成分冬青叶兔唇花(Lagochilusilicifolius)的谱系地理学进行了研究,发现在第四纪的气候波动中青藏高原快速隆升阻止了印度洋暖空气,同时加剧了冬季季风,使中国西北部持续干冷和沙漠化。对船苞翠雀花(Delphiniumnaviculare)谱系地理学的研究表明,更新世三次冰期-间冰期气候震荡过程对其地理分布格局和空间遗传结构产生重大影响,冰期时干旱和寒冷气候是造成其现代空间遗传结构的主要驱动力,并以阶梯式的扩张历史和等级式的避难所来响应干旱化的加强过程,并且在该地区形成的“干冷”气候使不同避难所之间的种群形成长期的空间隔离,进而促使不同种群间发生异域分化,导致异域式物种形成,为新种或新亚种形成提供重要条件[29]。分析当前中国西北部干旱区荒漠植物谱系地理学相关研究之后,初步得出中国西北部干旱区的干旱化过程促进了荒漠化的分化,而沙漠扩张导致了物种栖息地的碎片化,在新疆阿勒泰、天山地区及宁夏贺兰山等地存在冰期避难所,冰期后植物存在从避难所迁移和扩张的过程,以及在气候动荡时期,荒漠植物适应气候扩张而高山植物退缩[30-31]。
中国扁桃亚属共有6个种,分别是扁桃(Amygdaluscommunis)、矮扁桃(Amygdalusledebouriana)、长柄扁桃(Amygdaluspedunculata)、蒙古扁桃(Amygdalusmongolica)、西康扁桃(Amygdalustangutica)及榆叶梅(Amygdalustriloba)[32]。蒙古扁桃主要分布于内蒙古的鄂尔多斯西部、阿拉善中东部、贺兰山、甘肃的河西走廊等地。蒙古扁桃被中国植物物种信息数据库收录为国家珍稀濒危Ⅱ级重点保护植物,是西北干旱荒漠地区植被上的主要建群种,对维持该区脆弱生态系统的稳定具有重要作用[33]。蒙古扁桃在干燥石质低山形成灌木层片,不能形成大面积荒漠群落,只能在山地及沟谷形成局部片段的群落[34]。由于自然原因和人类经济活动特别是过度放牧、乱砍滥伐做燃料和大量采集其种仁榨油、入药,致使其数量锐减,处于濒危状态,野生种群面积逐渐缩小,且蒙古扁桃种子属核果类种子,其种壳坚硬落在缺水的沙地上难以发芽成苗,自然繁殖相当困难,种群数量快速下降,亟需对其进行濒危机制研究。同时蒙古扁桃为古地中海第三纪中新世孑遗落叶林树种,对研究古植物区系的变迁和古地理及第三纪以来的气候波动和环境变迁有着重要的科学价值。因此,本研究选取叶绿体DNA非编码区trnH-psbA序列对内蒙古、宁夏及甘肃的特有多年生灌木蒙古扁桃居群进行谱系地理学研究,已期揭示蒙古扁桃在居群内和居群间的遗传分布式样,单倍型的地理分布格局及其遗传结构;推断其在第四纪冰期避难所,与冰期后迁移路线,探讨蒙古扁桃形成现有分布格局的主要历史原因,为该物种种质资源的保护与利用和西北干旱区生态安全提供坚实的理论基础。
1 材料和方法
1.1 实验材料
研究材料采自蒙古扁桃分布的内蒙古、宁夏及甘肃等地区。共采集蒙古扁桃17个野生群落数据和居群的分子材料,共有324个个体(表1)。采集材料时居群内个体至少相隔10 m以上,采集新鲜叶片后用硅胶迅速干燥,实验室放入-20 ℃冰箱中保存,所采集的凭证标本存放于榆林学院植物标本馆。
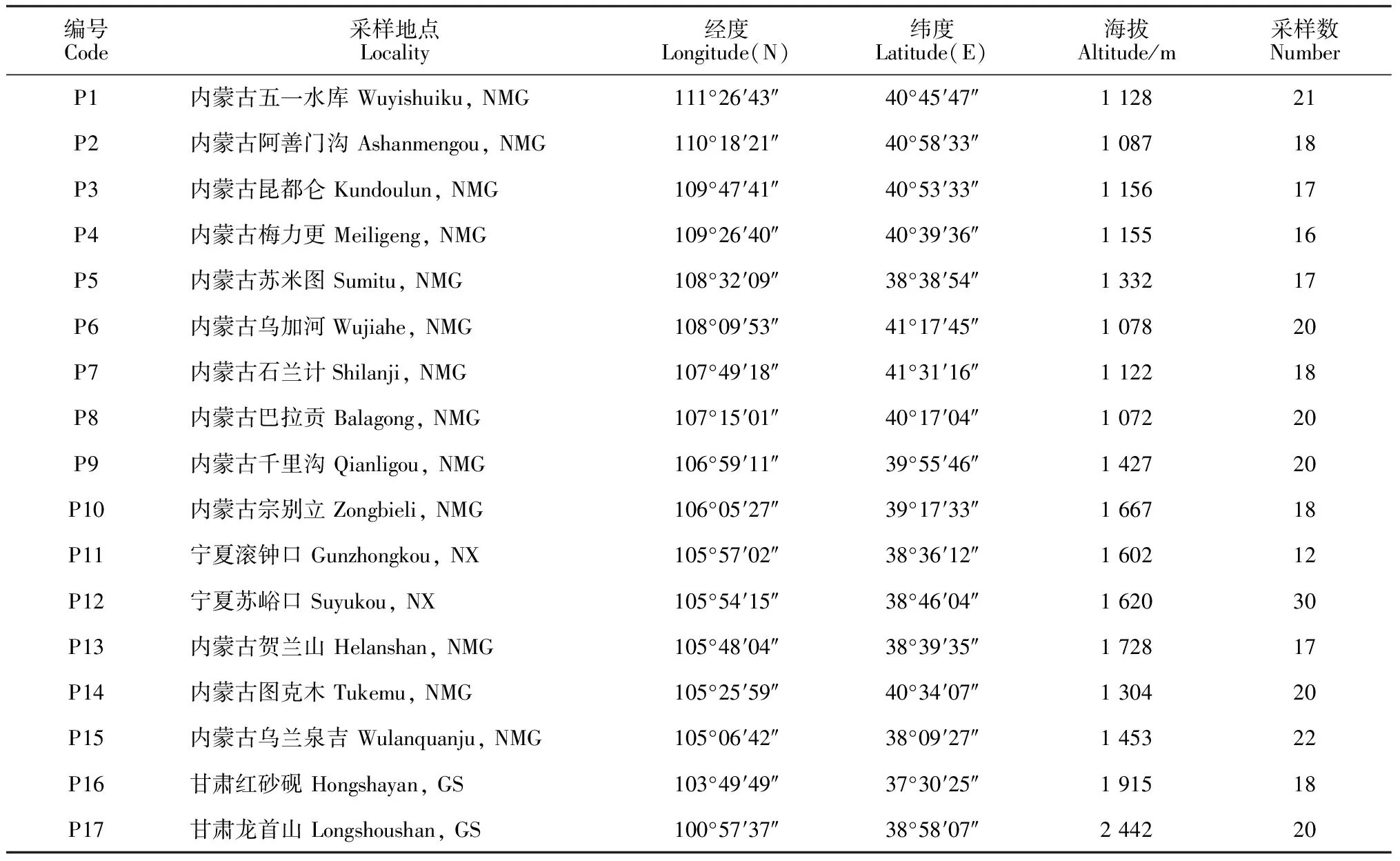
表1 蒙古扁桃17个居群的采样地点和个体数
注: NMG. 内蒙古自治区;NX. 宁夏回族自治区;GS. 甘肃省
Note: NMG. Inner Mongolia; NX. Ningxia Hui Autonomous Region; GS. Gansu Province
1.2 DNA的提取与PCR扩增
总DNA抽提利用柱式抽提纯化法试剂盒进行,用1%琼脂糖凝胶电泳检测,电泳结果显示为很亮的目的条带,质量良好。在对蒙古扁桃所有个体进行大规模扩增和测序之前,首先选择地理位置相隔较远的5个居群25个个体(每个居群5个个体)进行5个cpDNA非编码区trnS-trnG、trnH-psbA、rpl20-5′rps12、psbB-psbF以及trnL-trnF基因序列的扩增和测序,以寻找在种内居群水平上表现出较高的叶绿体非编码区序列。结果显示叶绿体trnH-psbA基因间隔区在蒙古扁桃种内居群水平上表现出了较高的变异,而其他基因间隔区种内居群水平上差异很小,因此确定cpDNA非编码区trnH-psbA基因序列用于蒙古扁桃谱系地理学研究。PCR扩增反应在Biometra thermals cycler PCR(TpersonaL 48)扩增仪上进行,PCR反应总体系为25 μL,包含2.5 μL 10×PCR缓冲液(MgCl2),1.6 μL 10 mmol/L dNTPs,0.15 μL Taq DNA聚合酶,各1 μL的正反trnH 及psbA引物,10 μL总DNA模板,8.75 μL灭菌蒸馏水;PCR反应程序为:94 ℃预热4 min,接着以94 ℃加热变性50 s,53.4 ℃低温退火30 s,72 ℃适温延伸90 s,循环34次,最后72 ℃延伸7 min。取2 μL PCR扩增产物进行1.0%琼脂糖凝胶电泳,检测扩增的产量和质量。成功扩增的PCR扩增产物采用Sunbiotech DNA 序列回收试剂盒(北京三博远志生物技术有限责任公司)进行纯化,将扩增产物直接送北京三博远志生物技术有限责任公司利用双向引物进行正反测序。
1.3 数据分析
测序所得的序列分别用Chromos软件进行正反链比对并手工进行校正,校对好的序列再用Clustal X软件排列,排完后适当手工校正,用DNASP 4.0软件统计变异位点并确定单倍型(Haplotype)。用MEGA 4.0.1软件计算序列的碱基组成、转换颠换比,用DNASP 4.0统计序列的总变异位点、简约信息位点及单突变位点。
应用软件ARLEQUIN 3.0 计算每个居群的单倍型多样性(He)、核苷酸多样性(π)。利用PERMUT软件计算居群内平均遗传多样性(Hs)、总的遗传多样性(Ht)、居群间遗传分化系数Gst和Nst值,使用U-统计方法对Gst和Nst进行比较(1 000次重复的置换检验),计算Gst值只使用了单倍型频率,代表了居群的分化程度,即居群间的遗传多样性占总遗传多样性的比例,可用公式Gst= (Hs-Ht)/Ht计算,而计算Nst还考虑了单倍型之间的差异。
居群遗传结构是遗传多样性在居群内和居群间的分布,即遗传分化,利用ARLEQUIN 3.0软件包中的AMOVA分析计算群体的遗传结构以及检测居群内和居群间遗传变异,并对居群内和居群间单倍型分布做Fst,以进一步揭示居群的分化程度(1 000次重复的显著性检验)。采用ARLEQUIN 3.0中对所有个体及每个居群进行歧点分布分析(mismatch distribution analysis)和中性检验(neutrality test)来检验蒙古扁桃居群是否经历了群体快速扩张事件。在居群近期突然扩张模型下,用1 000次参数自展重复来产生期望的歧点分布,计算适合度检验中的SSD值、R指数以及它们的显著性。同时用软件DNASP 4.0对所有个体进行歧点分布分析,观测到的歧点分布呈单峰曲线表示居群经历了近期扩张,而多峰曲线则表示在较长的时间内居群大小相对稳定,并处于个体平衡中。在本研究中,假设cpDNA变异处于漂变-迁移平衡,则物种水平上居群间的平均基因流Nm通过ARLEQUIN 3.0软件包检测得到的Fst值进行计算。
单倍型之间的网络关系通过软件Network 5.0构建。单倍型系统发育分析以近缘种榆叶梅(Peunustriloba,GenbBank登录号EU669088.1)为外类群,利用PAUP 4.0中的最大简约法(MP),最大似然法(ML),邻接法(NJ)分别进行。插入和缺失位点(indel)用程序GAPCODER设置为软件处理成为有或无状态(0或1)。PAUP参数设置如下:TBR 枝长交换,启发式搜索(Heuristic search),多重性选择MULPARS,ACCTRAN优化和100次随机附加的重复,用自展法(Bootstrap Analysis)检验系统树,自展数据集为1 000次。
2 结果与分析
2.1 单倍型多样性以及核苷酸多样性
蒙古扁桃 cpDNAtrnH-psbA序列成功测序17个居群共324个个体,经Clustal X软件对位排序后序列长度为350 bp, 其中(A+T)含量为81.5%,(C+G)含量为18.5%。应用DNASP 4.0软件对324个蒙古扁桃序列进行单倍型及变异位点统计,共有9个单倍型(图1,表2),有63处变异位点,变异位点的百分率为16.57%。在得到的63处变异位点,空位(gap)用作缺失处理,但其中只有3处为信息位点,空位及信息位点分别占总序列的18.00%和0.86%;3处信息位点位于139、228及300 bp处,出现2处碱基颠换(T/A),颠换率为3.44%,1处碱基转换(A/G),转换率为1.72%(表3)。
蒙古扁桃cpDNAtrnH-psbA序列检测到9种单倍型。其中单倍型H1的个体数为139个,频率最高;其次为单倍型H3的个体数为99个;单倍型H7及H9的个体数只有1个,频率最低。单倍型H1的地理分布范围也最广的,分布在8个居群,其中居群10、14、15、16及17只固定了单倍型H1;单倍型H3分布于7个居群中,其中居群2、3、5、8及9只固定了单倍型H3;特有单倍型分布范围狭窄,例如单倍型H6只存在于乌加河居群,单倍型H7、H8、H9只存在苏峪口居群内。

黄线表示划分的东西两个居群组,居群代号同表1图1 蒙古扁桃9种单倍型地理分布图The eastern group and western group of populations are divided by the yellow line, population code is the same as Table 1.Fig.1 Geographical distribution of the 9 haplotypes in A. mongolica
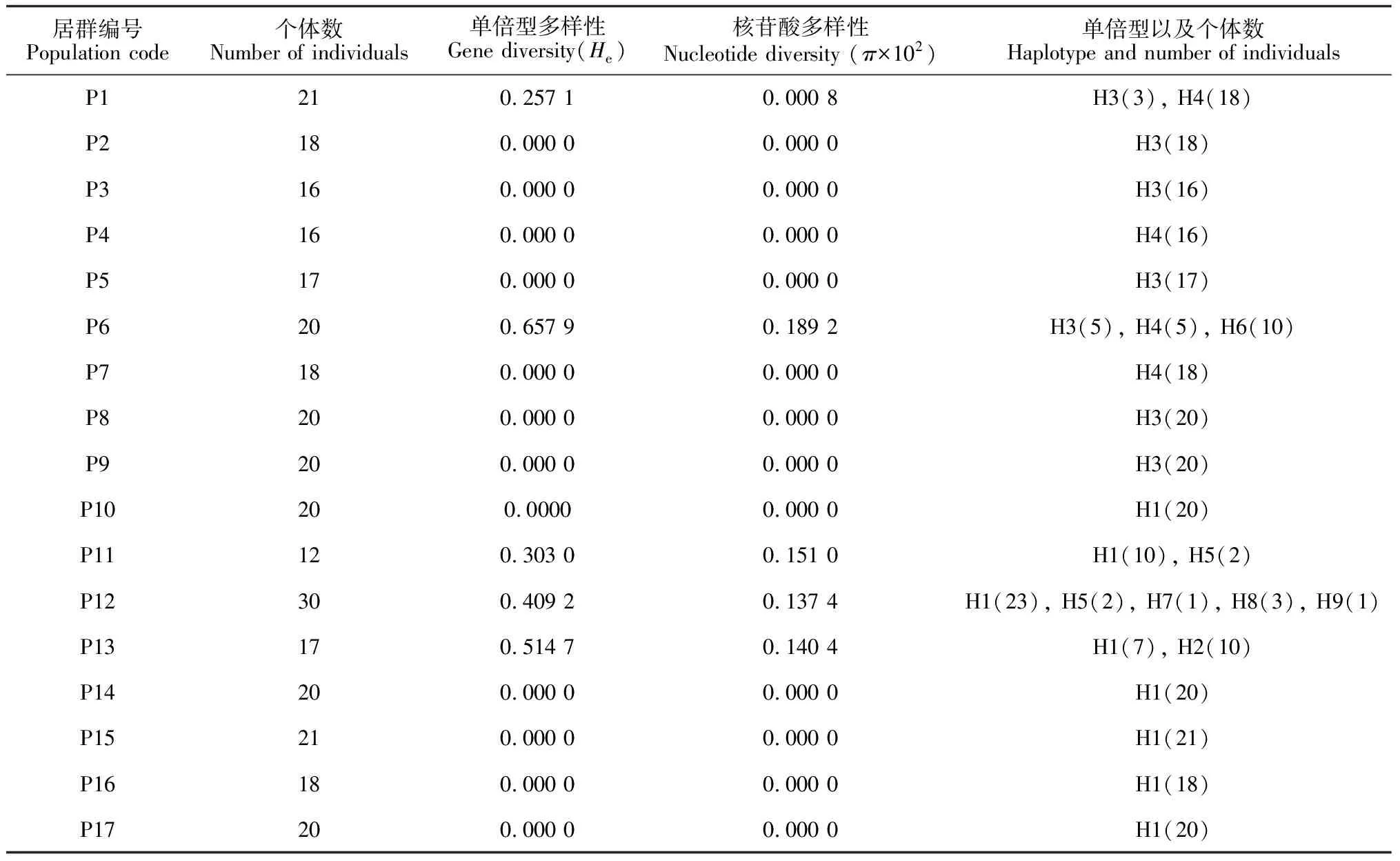
表2 蒙古扁桃17个居群的单倍型组成、单倍型多样性(He)及核苷酸多样性(π)
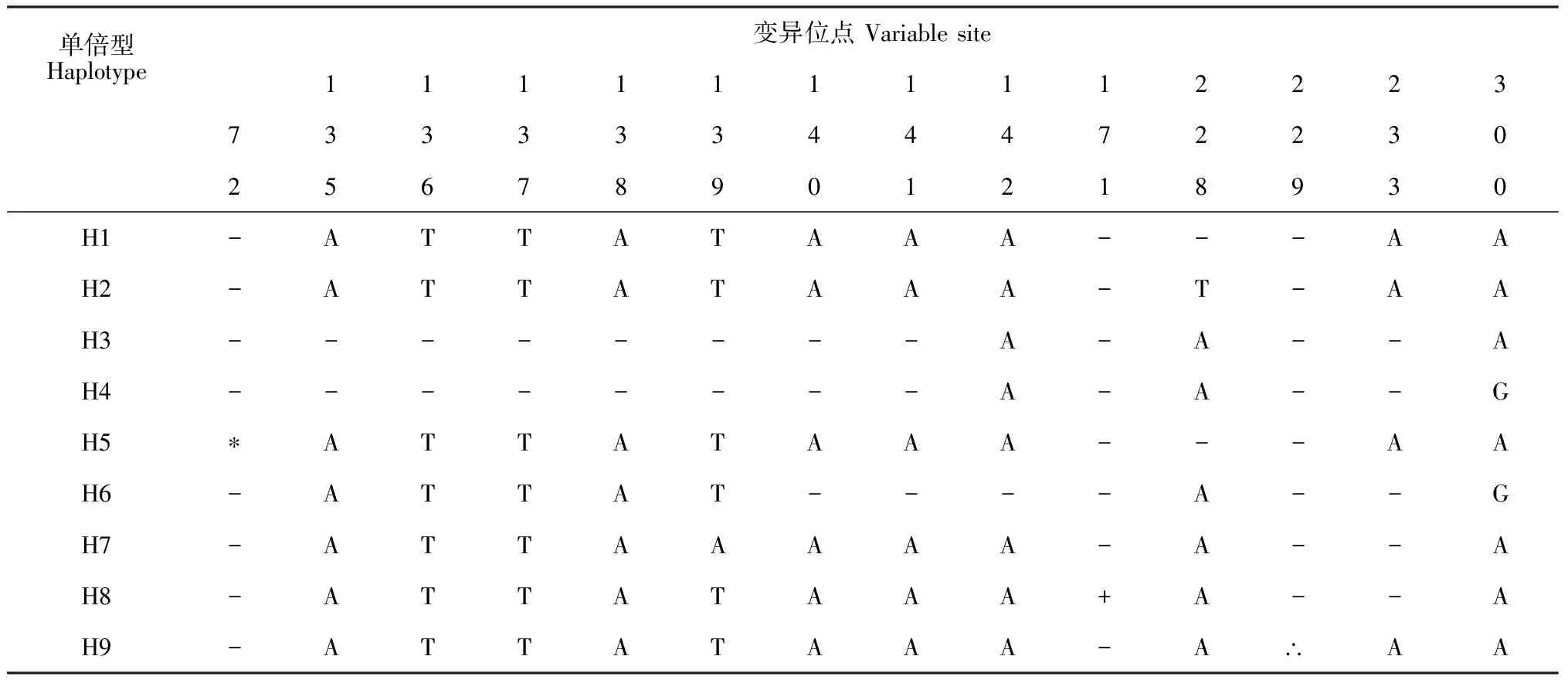
表3 蒙古扁桃cpDNA trnH-psbA序列变异位点
注:缩写,*. AAGATAAAATACAACATAAAATAAACT; +. TAGTAAAAATATAGAACAATA; ∴ CTAA; - 缺失的碱基
Note: *. AAGATAAAATACAACATAAAATAAACT; +. TAGTAAAAATATAGAACAATA; ∴ CTAA; - indel
由表2可知,在蒙古扁桃17个居群中,单倍型多样性(He)的范围从0到0.657 9,其中采自乌加河居群的单倍型多样性最高(0.657 9),苏峪口居群的单倍型单倍型次之(0.409 2);而核苷酸多样性范围是从0到0.189 2,乌加河居群具有最高的核苷酸多样性(0.189 2),其次为滚钟口的居群(0.151 0)。在蒙古扁桃整个分布区,通过cpDNAtrnH-psbA序列9种单倍型的地理分布格局可以看出以贺兰山-阴山作为分界线(图1),以东的居群固定单倍型H3、H4及H6,多数居群只固定了一个单倍型,只有位于阴山南麓的乌加河居群固定了3种单倍型,且核苷酸多样性也较高;以西的居群固定的单倍型有H1、H2、H5、H7、H8及H9,西部居群单倍型数量较东部居群多,尤其在贺兰山东麓的苏峪口及滚钟口居群固定了大量的特有单倍型。值得注意的是东部的居群与西部的居群不存在共享单倍型(图1)。
2.2 居群遗传结构分析
利用PERMUT软件对蒙古扁桃cpDNAtrnH-psbA序列的9种单倍型进行计算。结果表明,居群内平均遗传多样性(Hs)为0.203,总的遗传多样性(Ht)为0.758,居群间遗传分化Gst和Nst分别为0.733和0.655,结果为Nst小于Gst,(P>0.05),表明在整个分布区不存在明显的分子谱系地理学结构。
2.3 蒙古扁桃cpDNA trnH-psbA序列分子变异分析
根据单倍型地理分布位置及Network单倍型网络关系(图2),把蒙古扁桃整个地理分布区分为东部居群组(P1~P9)与西部居群组(P10~P17)。利用ARLEQUIN软件包对蒙古扁桃居群进行分子变异分析(AMOVA),结果(表4)表明蒙古扁桃cpDNAtrnH-psbA序列的遗传变异居群间显著大于居群内,居群间的遗传变异为83.84%,居群内遗传变异为16.16%;对于东,西居群组而言,有84.72%的遗传变异来源于组间,5.55%的遗传变异来源于组内居群间,9.73%的遗传变异来源于居群内;总的居群间及东、西居群组间的Fst值为分别为0.84及0.90(P<0.001,1 000次重复的显著性检验)(表4)。假设该序列变异处于漂变-迁移平衡(drift-migration equilibrium),则基于Fst值估算出物种水平上居群间的平均基因流值(Nm)为0.10。
2.4 蒙古扁桃cpDNA trnH-psbA单倍型网络关系分析
基于Network 5.0中邻接法构建的蒙古扁桃单倍型网络关系(图2)表明,单倍型H1位于网络图的中央位置,推测其可能是最古老的单倍型,分布在东部居群组的单倍型H3、H4及H6聚为一支,分布在西部居群组的单倍型H8及H9聚为一支,而单倍型H2、H5及H7各自单独成支(图2)。利用PAUP 4.0中的最大简约法(MP),最大似然法(ML),邻接法(NJ)构建蒙古扁桃系统发育树支持率均较低,可能的原因在于蒙古扁桃单倍型之间存在大量的插入及缺失的碱基,碱基的替换只有3处,因此系统发育树支持率均较低。
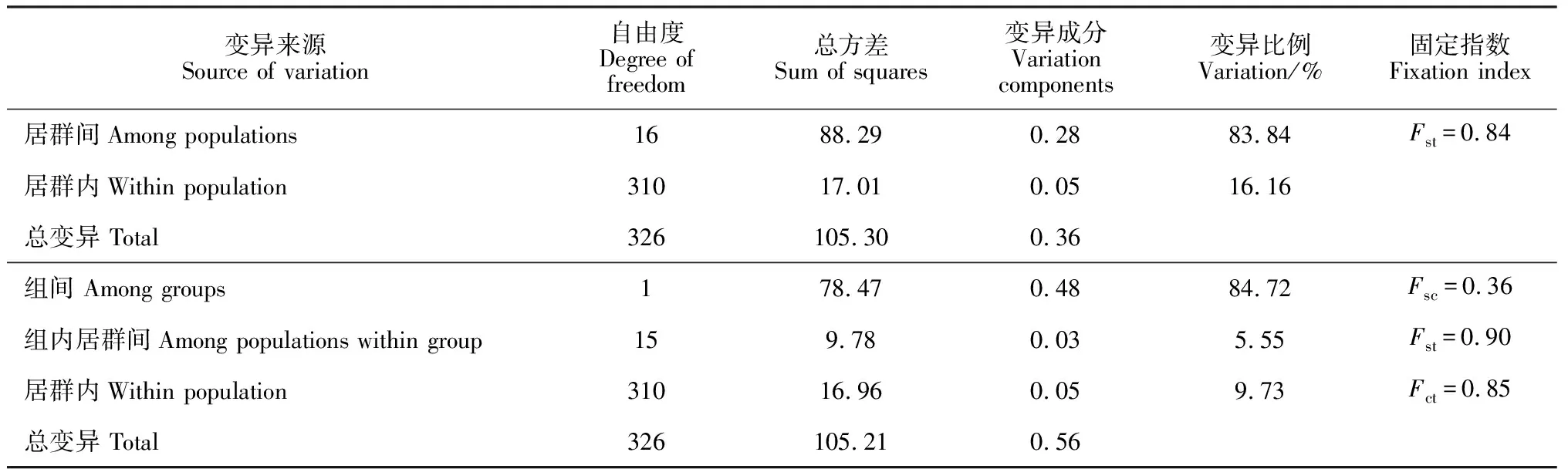
表4 蒙古扁桃17个居群的分子变异分析
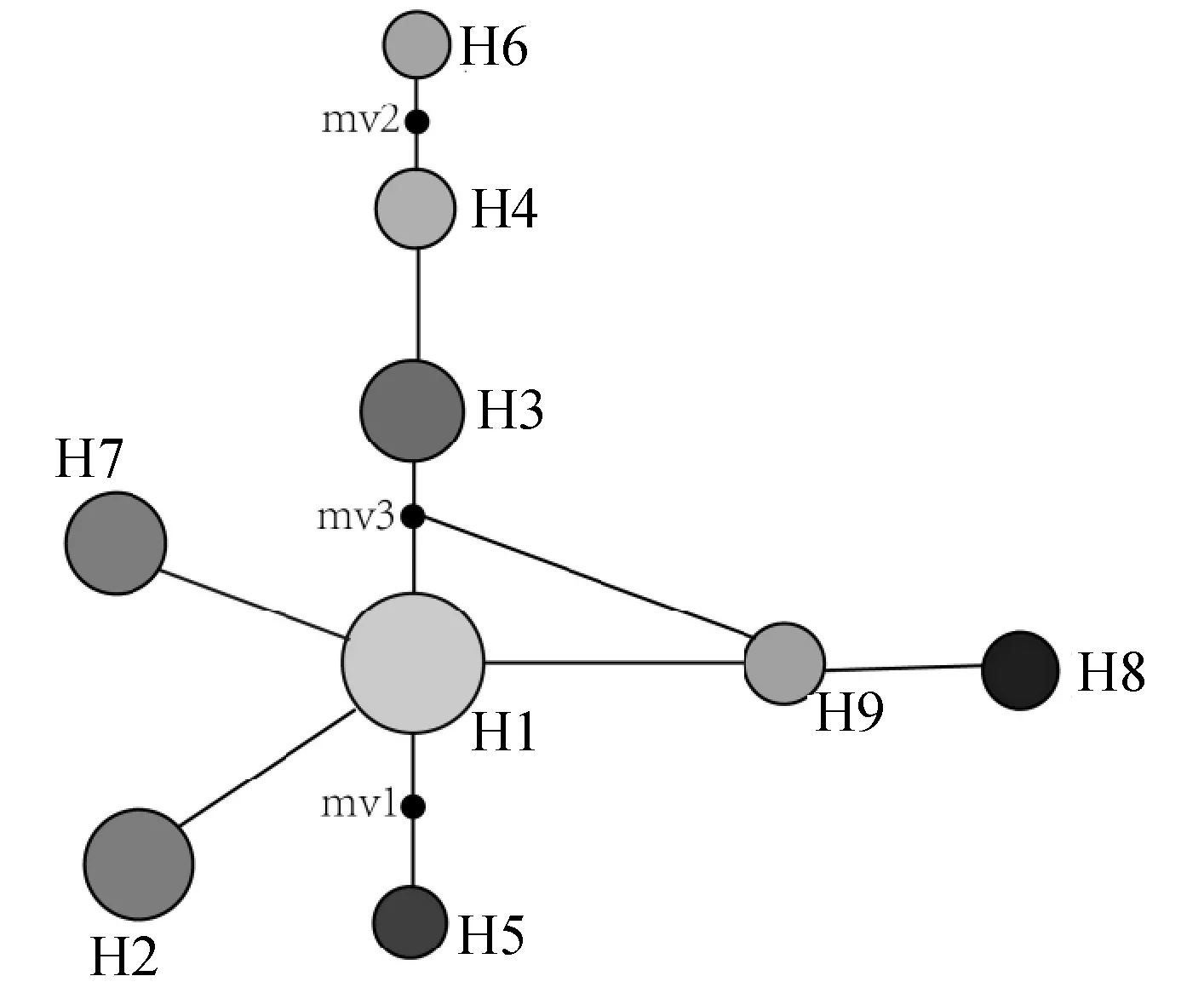
图2 蒙古扁桃基于trnH-psbA序列数据单倍型网络Fig.2 Haplotype network of A.mongolica based on trnH-psbA sequence
2.5 蒙古扁桃cpDNA trnH-psbA序列居群近期扩张分析
蒙古扁桃17个居群324个个体trnH-psbA序列数据的3种无限突变位点模型中性检验值,结果均为负值:Tajima’sD: -0.317,Fu and Li’sD* test statistic: -0.504,Fu and Li’sF* test statistic: -0.524,但不显著(P>0.10);此外,歧点分布(Mismatch distribution)分析结果如图3所示,观测值与期望值之间比较,得到了一个很明显的单峰分布曲线图,上述结果表明蒙古扁桃经历过居群近期扩张的过程。
3 讨 论
3.1 冰期避难所推测
应用DnaSP4.0软件对蒙古扁桃trnH-psbA序列的全部个体进行单倍型统计,初步鉴定出11单倍型[35],其中4种单倍型只出现1次,通过对原始序列校正后发现,其中2种单倍型变异倍点存在一定的套峰现象,推测它们产生差异的原因是由于测序技术导致的,并不是个体自身存在差异,因此将这2种单倍型去除,最终鉴定出9种单倍型。单倍型网络关系显示单倍型H1位于网络中心位置,推测其为最古老的单倍型。基于单倍型地理分布位置及网络关系,把蒙古扁桃自然地理居群分为东、西2大地理组群。西部组群中,在贺兰山东麓的苏峪口及滚钟口的居群内都包含单倍型H1,还有4种特有单倍型,而且这3个地区居群的单倍型多样性及核苷酸多样性均较高,因此推测贺兰山为蒙古扁桃西部组群的冰期避难所;东部组群中,位于阴山南麓的乌加河居群包含东部组群所有的3种单倍型,这其中有1种为特有单倍型,该居群单倍型多样性及核苷酸多样性在17个居群中最高,因此推测阴山南麓的乌加河为蒙古扁桃东部组群的冰期避难所。造成贺兰山东麓及阴山南麓蒙古扁桃单倍型多样性和核苷酸多样性高于其他地区居群的可能原因是由于这2个地区的高大山脉阻碍了沙漠化的扩张,导致这一地区的温度比同期冰期中其他地区的温度要略高,而这些残留的蒙古扁桃居群在冰期中并没有发生迁移或退缩,仍然留在原地,因而这2个地区成为蒙古扁桃在第四纪冰期导致沙漠化扩张以及寒冷期间的避难场所。
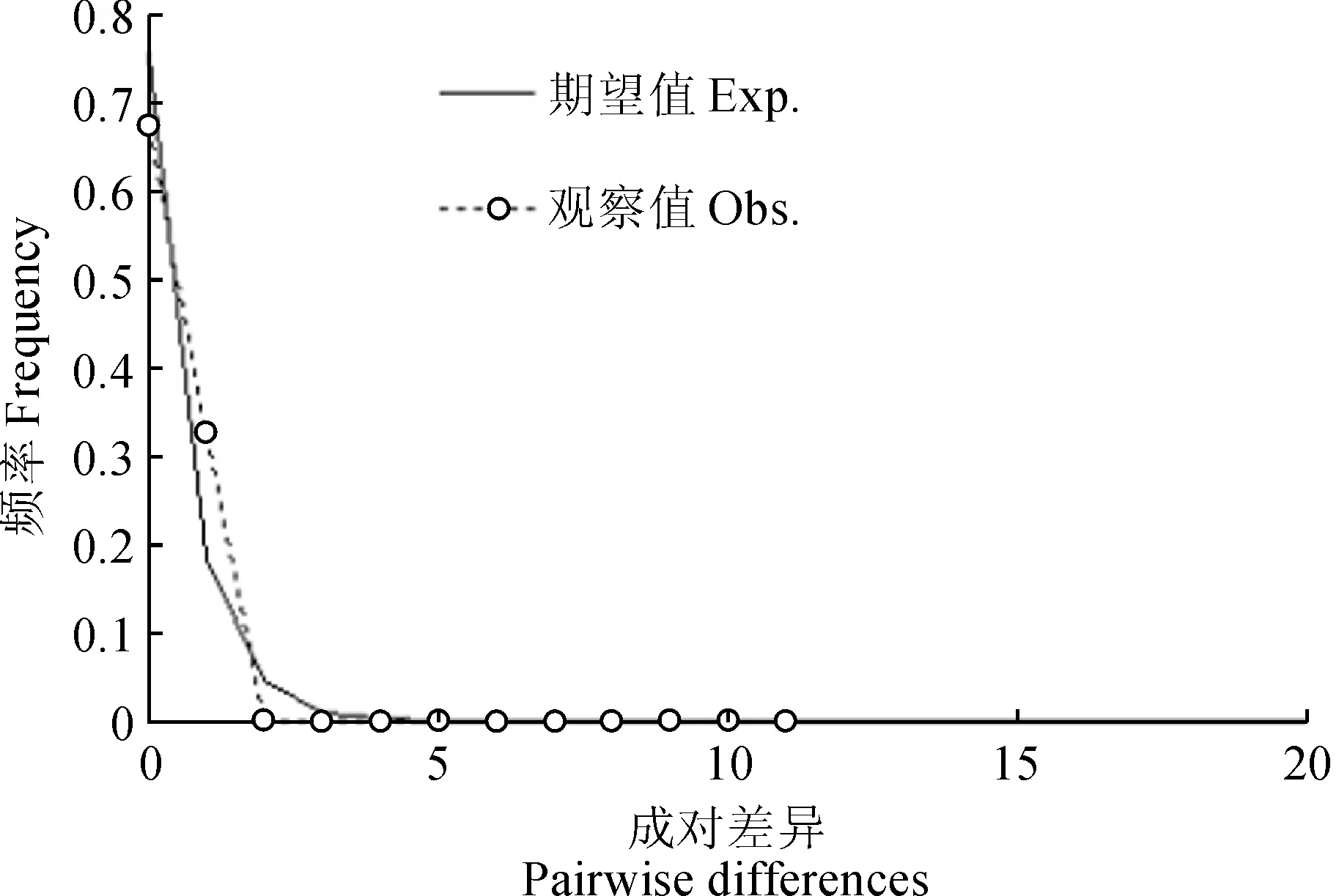
图3 蒙古扁桃324个个体trnH-psbA序列数据的歧点分布图Fig.3 Mismatch distribution for sequence data of the trnH-psbA sequence from 324 individuals of A.mongolica
3.2 群体遗传结构与分化
蒙古扁桃东、西2大地理组间存在明显的遗传分化(84.72%,P<0.001),西部组群包括的单倍型有H1~H2、H5、H7~H9,东部组群包括的单倍型有H3、H4及H6,2个组群之间无共享单倍型。分子变异分析及Permut分析表明,蒙古扁桃居群间的遗传分化水平较高(Gst=0.733),蒙古扁桃总的遗传多样性也较高(Ht=0.758),造成蒙古扁桃东、西组群地理居群遗传分化的原因可能是由于基因流较低,地理隔离及种子传播距离有限等因素。具体来说,基于Fst值估算出蒙古扁桃居群间的平均基因流值(Nm)为0.10,基因流较低;蒙古扁桃种子粒径较大,重量较沉,因此种子长距离传播能力有限;加之有黄河等河流及沙漠、山脉等地理隔离因素阻隔了蒙古扁桃东西部自然地理居群基因交流,使得东西部居群出现了明显的遗传分化。此外,居群的遗传分化水平与蒙古扁桃的异花授粉或虫媒传粉有关系。
3.3 居群历史动态分析
基于Tajima’sD,Fu and Li’sD,Fu’s DF中性检验值及其错配分布分析表明了蒙古扁桃居群经历了近期的居群扩张。因蒙古扁桃为第三纪孑遗植物,进而推测蒙古扁桃在第四纪冰期分别在贺兰山东麓及阴山南麓避难所保存下来,冰期结束后从避难所分别向外扩张,伴随着奠基者效应的发生,加之冰期与间冰期的反复交错,居群将会经历一系列的瓶颈效应以及强烈的生境片段化,使得迁移的居群遗传多样性降低,致使绝大部分居群只固定了一种单倍型。根据单倍型网络关系图推测H1为最古老的单倍型,其他单倍型都是由单倍型H1演化而来,西部组群中每个居群都含有单倍型H1,表明西部居群有着更长的进化历史,而东部组群中只有由单倍型H1演化而来的H3、H4及H6,表明东部组群进化历史较西部组群短。
综上所述,基于cpDNAtrnH-psbA序列对蒙古扁桃17个居群324个个体进行谱系地理学研究表明,蒙古扁桃在第四纪冰期的避难所存在于贺兰山东麓及阴山南麓,造成蒙古扁桃东西部自然地理居群出现明显的遗传分化主要是由于居群间基因流较小,地理隔离及种子长距离传播能力有限等原因。蒙古扁桃的保护价值不仅在于其具有观赏和药用价值,更重要的它是国家珍惜濒危Ⅱ级重点保护植物[35]。对蒙古扁桃居群的保护首先要考虑避难所一带的居群即贺兰山东麓及阴山南麓地区的居群,除采取原地保护策略外,还应采取迁地保护,迁地保护的优点是快捷方便,避免物种灭绝,有效防止生物遗传多样性的丧失,弥补就地保护的一些不足,是就地保护的补充,操作相对简单经济。
