超高效液相色谱-四极杆飞行时间质谱法快速筛查水产品中16种激素残留
2018-10-31陈秋华张天闻黄东仁
陈秋华,张天闻*,傅 红,3,黄东仁
(1.福州大学生物科学与工程学院,福建 福州 350108;2.福建省海洋环境与渔业资源监测中心,福建 福州 350003;3.福州大学 福建省海洋酶工程重点实验室,福建 福州 350108)
激素是指由体内的某一细胞、腺体或者器官所产生的可以影响机体内其他细胞活动的化学物质,主要包括性激素、孕激素和糖皮质激素等[1]。激素具有提高饲料转化率、缩短动物生长周期、促进动物生长繁殖等作用,因此存在在水产养殖中违规使用以提高经济效益的现象[2-5]。研究表明,食品中激素的残留会导致性早熟、乳腺癌及前列腺癌发病率提高[6-7]。2002年我国农业部第235号公告中已禁止使用诺龙、甲基睾丸酮、群勃龙等激素物质,并规定这些化合物在动物性食品中不得检出[8]。同时欧盟第96/22/EC指令、美国食品药物管理局也禁止在动物源性食品中使用激素类药物[9]。
目前激素类药物的残留检测方法主要有高效液相色谱法[10]、液相色谱-串联三重四极杆质谱法[11-14]、超高效液相色谱-四极杆飞行时间质谱法[15-18]等。其中液相色谱法无法区分保留时间相同或相近的物质,因而定性能力较差。液相色谱-串联三重四极杆质谱法凭借多反应监测模式,其灵敏度和定性能力都较高,是目前主要的检测方法,但受分辨率、扫描速率及分析模式的限制,无法实现高通量快速筛查分析[19]。液相色谱-四极杆飞行时间质谱法具有高分辨率、高通量、灵敏度高的特点,可实现复杂食品基质中目标物的定性分析和高通量快速筛查分析,同时还可通过建立筛查谱库实现在无标准品情况下对目标物进行筛查和确认[20-23]。
QuEChERS(Quick,Easy,Cheap,Effective,Rugged and Safe)是由Anastassiades等[24]于2003年建立的样品前处理技术,具有操作简单、分析速度快、分析范围广等优点,此方法已广泛应用于食品中农药残留的检测,近年来也逐渐被用于动物性食品中兽药残留的检测[25-27]。熊雯等[28]建立了检测水产品中18 种磺胺类药物的QuEChERS-液相色谱-四极杆飞行时间质谱方法。朱万燕等[29]建立了快速检测猪肉中33 种兽药残留的QuEChERS-液相色谱-四极杆飞行时间质谱方法。但目前采用QuEChERS-液相色谱-四极杆飞行时间质谱方法同时对多种水产品基质中的激素残留进行筛查分析的文献报道还较少。本实验通过超高效液相色谱-四极杆飞行时间质谱(ultra performance liquid chromatographyquadrupole time of fl ight mass spectrometry,UPLC-Q-TOF MS)技术建立筛查数据库,同时采用改进的QuEChERS方法结合UPLC-Q-TOF MS技术建立水产品中激素快速、有效的筛查方法,以期为水产品中激素残留风险监测提供有效技术手段,同时为水产品中兽药残留的筛查分析提供借鉴。
1 材料与方法
1.1 材料与试剂
勃地酮、甲睾酮、睾酮、诺龙、美雄酮、表睾酮、群勃龙、17α-羟基孕酮、甲羟孕酮乙酸酯、乙酸氯地孕酮、21α-羟基孕酮、乙酸甲地孕酮、醋酸美伦孕酮、孕酮、氢化可的松、泼尼松、睾酮-D3(纯度均大于93.6%) 德国Dr. Ehrenstorfer公司;甲醇、乙腈、乙酸乙酯、正己烷、丙酮(均为色谱纯) 德国Merck公司;甲酸(色谱纯) 美国Scientific公司;乙酸(色谱纯)美国Tedia公司;无水硫酸镁(分析纯) 国药集团化学试剂有限公司;氯化钠(农残级) 上海安谱公司。
1.2 仪器与设备
ACQUITY H-CLASS型UPLC仪、Xevo G2-S型Q-TOF MS仪 美国Waters公司;V-700旋转蒸发仪德国Heidolph公司;3-30K高速冷冻离心机 德国Sigma公司;MS3 digital旋涡振荡器 德国IKA公司;multi Reax振荡器 德国Heidolph公司;Milli-Q超纯水系统美国Millipore公司。
1.3 方法
1.3.1 标准溶液的配制
分别准确称取适量标准物质或同位素内标物质,用甲醇溶解并定容至100 mL,配制成100 μg/mL的单标储备液,避光保存于-18 ℃冰箱。准确移取适量标准储备液用甲醇定容为500 ng/mL的混合标准储备液,保存于4 ℃冰箱。用甲醇-水体积比35∶65稀释混合标准储备液获得1、5、10、25、50、100 μg/L的标准工作液,其中标准工作液中均含有20 μg/L的同位素内标睾酮-D3,将标准工作液保存于4 ℃冰箱。
1.3.2 样品前处理
准确称取2.00 g样品(精确至0.01 g)于50 mL离心管中,加入80 μL 500 ng/mL的激素混标溶液,静置15~20 min,加入10 mL 1%乙酸-乙腈溶液,涡旋混匀1 min,加入盐析剂(2 g无水硫酸镁和1 g氯化钠),涡旋混匀1 min,在4 ℃、10 000 r/min离心5 min,准确吸取上清液5 mL于新的15 mL离心管中,加入3 mL正己烷,涡旋混匀1 min,在4 ℃、5 000 r/min离心5 min,弃去正己烷层。再往离心管中加入净化剂(800 mg无水硫酸镁和150 mg C18(南美白对虾、大黄鱼、草鱼基质)或200 mg C18(梭子蟹基质)),涡旋混匀2 min,在4 ℃、5 000 r/min离心10 min,准确移取3 mL上清液于25 mL鸡心瓶中。38 ℃旋蒸至干,用1 mL含20 ng/mL睾酮-D3的定容液(甲醇-水体积比35∶65)复溶,过0.22 μm有机滤膜,待上机测定。
1.3.3 UPLC条件
色谱柱:Waters Acquity UPLC BEH C18柱(3.0 mm×100 mm,1.7 μm);柱温40 ℃;样品室温度10 ℃;进样体积10 μL;流动相:A为0.1%甲酸溶液,B为甲醇;流速0.3 mL/min;梯度洗脱条件:0~1 min,35% B;1~5 min,35%~65% B;5~8 min,65%~80% B;8~11 min,80%~98% B;11~14 min,98% B;14~15 min,98%~35% B;15~18 min,35% B。
1.3.4 MS条件
离子源:电喷雾离子源;电离模式:正离子;检测模式:MSE;质量数采集范围:50~1 000 Da;毛细管电压:3 kV;离子源温度:120 ℃;脱溶剂气温度:420 ℃;脱溶剂气流速:800 L/h;锥孔气流速:50 L/h;锥孔电压:40 V;LockSpray程序:采用亮氨酸脑啡肽(1 μg/mL,m/z 556.277 1)每10 s切换一次对质量数进行实时校正。
1.3.5 数据库的建立
本实验在MSE全扫描模式下,在低能量碰撞通道获得目标物的保留时间、母离子精确质量数等信息,在高能量碰撞通道获得目标物的子离子精确质量数。在MS/MS模式下,通过相应碰撞能量下的离子碎片信息进一步确证。借助Waters公司Masslynx 4.1软件中的fragment软件分析化合物的结构裂解信息。最后通过Chromalynx xs数据库软件,建立包含16 种激素名称、分子式、保留时间、精确质量数、子离子等信息的筛查数据库。具体的数据库信息如表1所示,色谱图见图1。
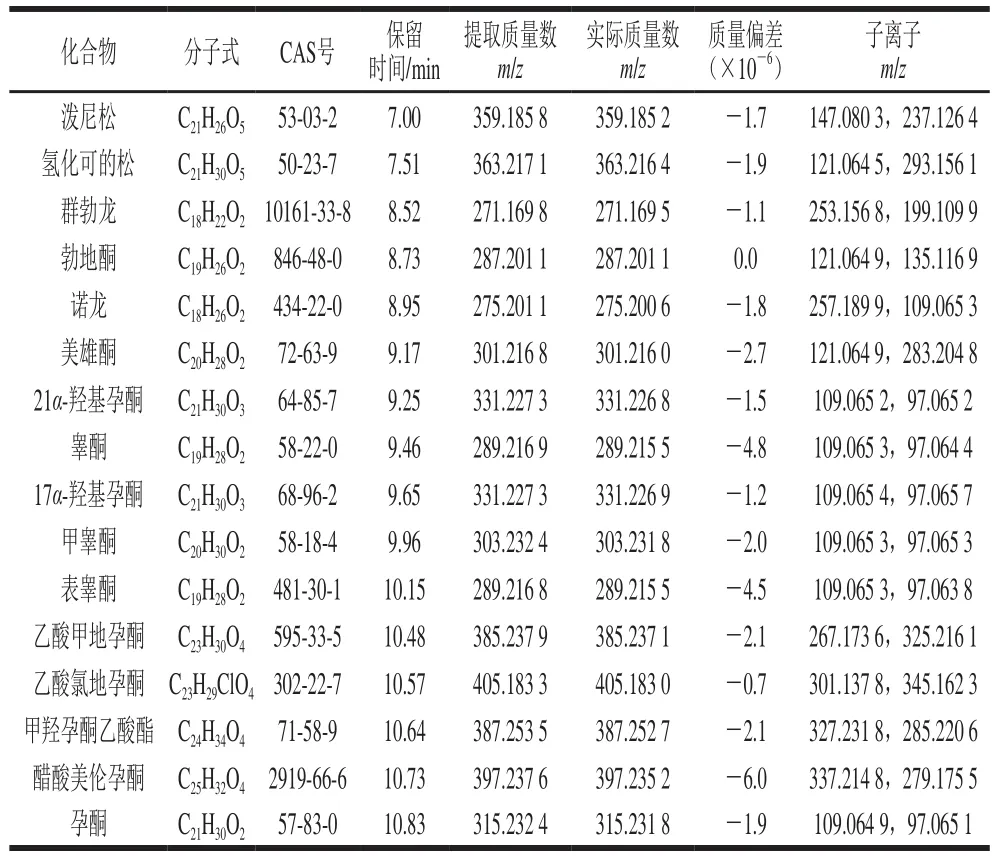
表1 激素的分子式、CAS号、保留时间、提取质量数、子离子Table 1 Molecular formula, CAS No., retention time, m/z and product ion of 16 hormones
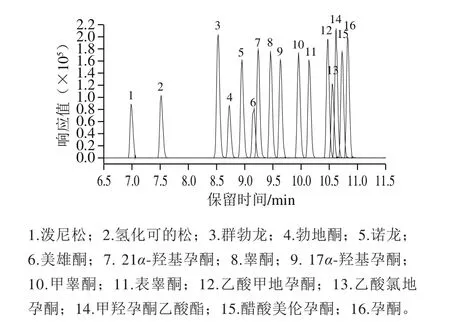
图1 16 种激素的色谱提取图Fig. 1 Extracted ion chromatogram of 16 hormones
1.3.6 基质效应的计算
基质效应的消除方法主要有内标法、基质匹配等。基质效应是通过比较目标化合物在纯溶剂与在基质中的峰面积进行评估的,其计算公式如下:

1.4 数据处理
在Chromalynx xs软件中,运用建立好的数据库对UPLC-Q-TOF MS采集的样品数据进行检索匹配分析,利用质谱软件的自动去卷积功能,并与数据库的保留时间、精确母离子质量数、子离子质量数等相关参数进行对比,当分析物与谱库中化合物的保留时间偏差小于±0.2 min,母离子和子离子质量数偏差小于±3 mu时,显示为阳性结果,随后针对阳性化合物使用targetlynx软件进行定量分析。与MS/MS相比,Q-TOF具有更快的扫描速度和更高的质量精度,从而可实现高通量筛查,另外本方法构建的是一个开放的体系,待测样品一次进样,经Q-TOF全扫描获得的全谱数据可多次使用,后期可通过不断增加数据库中的化合物来扩展对原样品数据的筛查范围。
2 结果与分析
2.1 MS条件优化结果
为获得最佳的电离效率、提高灵敏度,本实验以100 μg/L的混合标准溶液对锥孔电压、离子源温度、脱溶剂气温度、脱溶剂气流速等质谱参数进行优化,根据总离子流响应值和各化合物的分子离子峰响应值的强度确定最佳质谱参数。以锥孔电压为例,锥孔电压太小会降低检测灵敏度,太大则会造成离子源内裂解,本实验研究了16 种激素在锥孔电压为30~100 V时分子离子峰的响应情况,实验发现当锥孔电压为42 V时,激素总体响应值最高,同理对质谱的离子源温度(100~150 ℃),去溶剂化温度(350~500 ℃)和脱溶剂气流速(500~1 000 L/h)进行优化,优化后的质谱参数见1.3.4节。
2.2 QuEChERS样品前处理的优化
2.2.1 提取方法的优化结果
本实验考察乙腈、1%乙酸-乙腈溶液、乙酸乙酯和丙酮4 种提取剂的效果。如图2所示,分别用南美白对虾、大黄鱼和梭子蟹为基质,1%乙酸-乙腈溶液作为提取剂时16 种激素的平均回收率分别为83.3%、81.4%和83.8%,均高于乙腈、乙酸乙酯和丙酮,因而本实验选用1%乙酸-乙腈溶液作为提取溶剂。实验中还发现,乙酸乙酯和丙酮作为提取剂时,共提物较多,增加了提取液后期的净化难度,有乙腈存在的体系中共提物较少,这可能因为乙腈具有沉淀蛋白质的作用,有利于后期的净化。
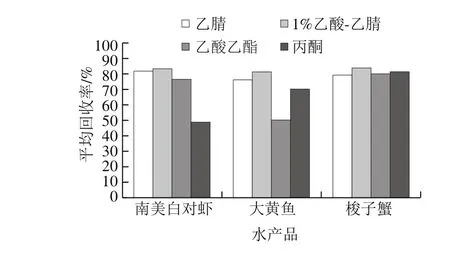
图2 不同提取剂对16 种激素回收率的影响Fig. 2 Effects of different extraction solvents on the recoveries of 16 hormones
2.2.2 净化方法的优化结果
QuEChERS方法常用的吸附剂有乙二胺-N-丙基硅烷(primary secondary amine,PSA)、十八烷基键合硅胶吸附剂(C18)、中性氧化铝(Alu-N)、石墨化碳黑(graphitized carbon blacks,GCB)等,其中C18和Alu-N主要用于去除样品中的脂肪、脂类等非极性有机物,而PSA和GCB主要用于去除样品中的色素、有机酸和糖类等物质。本实验分别考察以PSA、C18、Alu-N以及PSA-Alu-N、PSA-C18、PSA-C18-GCB作为净化剂时的净化效果,南美白对虾、大黄鱼、梭子蟹基质的净化效果如图3~5所示。实验结果表明,以C18吸附剂作为净化剂时16 种激素净化效果最好,在南美白对虾、大黄鱼和梭子蟹基质中,其回收率分别在61.9%~93.9%、64.2%~119.7%、55.6%~112.5%范围内。因此本实验采用选择C18作为吸附剂。本实验还进一步对C18的用量进行优化,对比了100、150、200、250、300 mg C18吸附剂对目标化合物回收率的影响,实验结果如图6所示,在南美白对虾、大黄鱼和梭子蟹基质中,C18吸附剂加入量分别为150、150、200 mg时净化效果最好,16 种激素平均回收率分别为83.5%、85.6%、84.0%。其中C18吸附剂加入量过低或过高都会使回收率降低,这可能是由于C18吸附剂加入量不够时,无法净化充分;而加入量过多时,吸附剂本身对激素也存在一定的吸附作用。
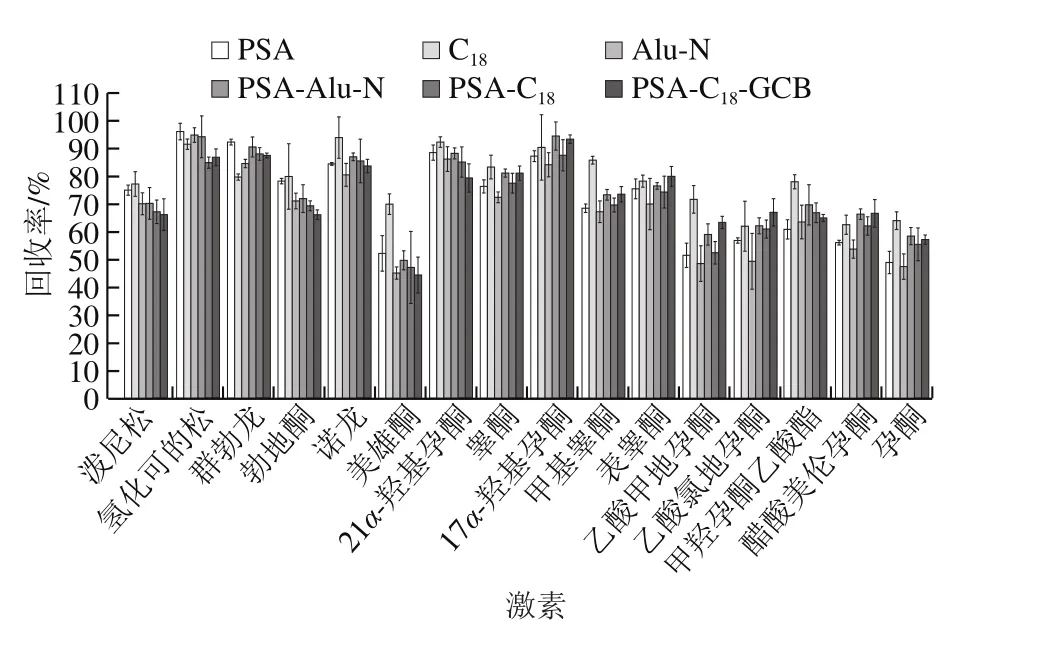
图3 南美白对虾基质中不同吸附剂对16 种激素的回收率的影响Fig. 3 Effects of different sorbents on the recoveries of 16 hormones in Penaeus vannamei matrix
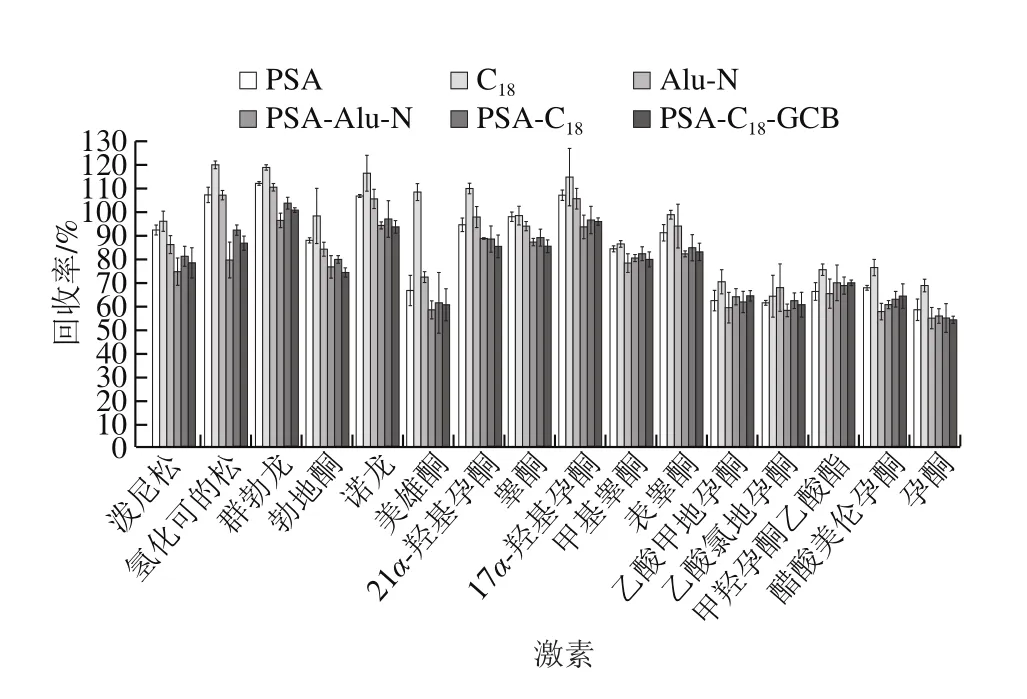
图4 大黄鱼基质中不同吸附剂对16 种激素的回收率的影响Fig. 4 Effects of different sorbents on the recoveries of 16 hormones in Pseudosciaena crocea matrix
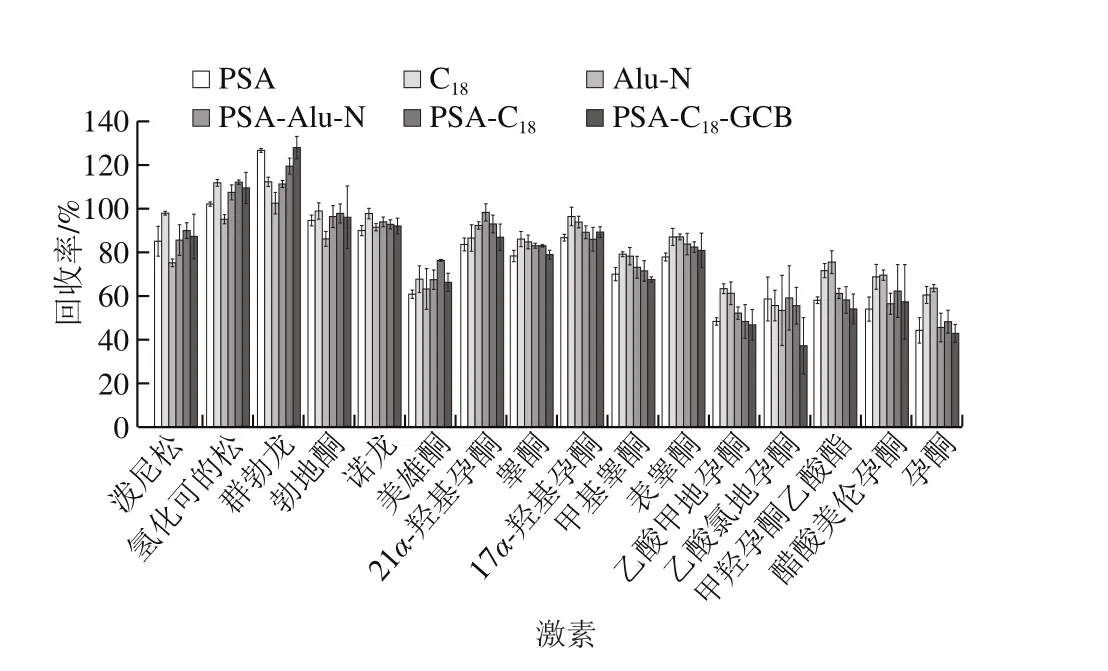
图5 梭子蟹基质中6 种吸附剂对16 种激素的回收率的影响Fig. 5 Effects of different sorbents on the recoveries of 16 hormones in swimming crab matrix
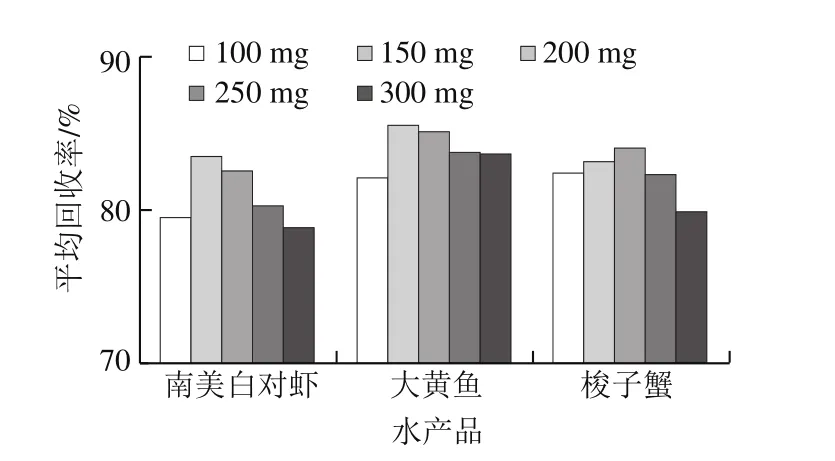
图6 C18吸附剂添加量对16 种激素回收率的影响Fig. 6 Effects of C18 amount on the recoveries of 16 hormones
2.3 方法学验证结果
2.3.1 基质效应
当基质效应在-20%~20%范围内时,基质效应不明显,超过20%时,表现为明显的基质增强效应,低于-20%时,表现为明显的基质抑制效应[30]。如表2所示,在南美白对虾基质中,16 种激素的基质效应均在±20%范围内,基质效应不明显;在大黄鱼基质中,勃地酮、美雄酮、泼尼松表现为明显的基质抑制效应;在梭子蟹基质中,美雄酮表现为明显的基质抑制效应,氢化可的松表现为明显的基质增强效应;在草鱼基质中,勃地酮、美雄酮、表睾酮、泼尼松表现为明显的基质抑制效应。为获得更准确的结果,本实验采用通过基质匹配溶液的方法对基质效应进行校正。
2.3.2 线性范围、检出限和定量限
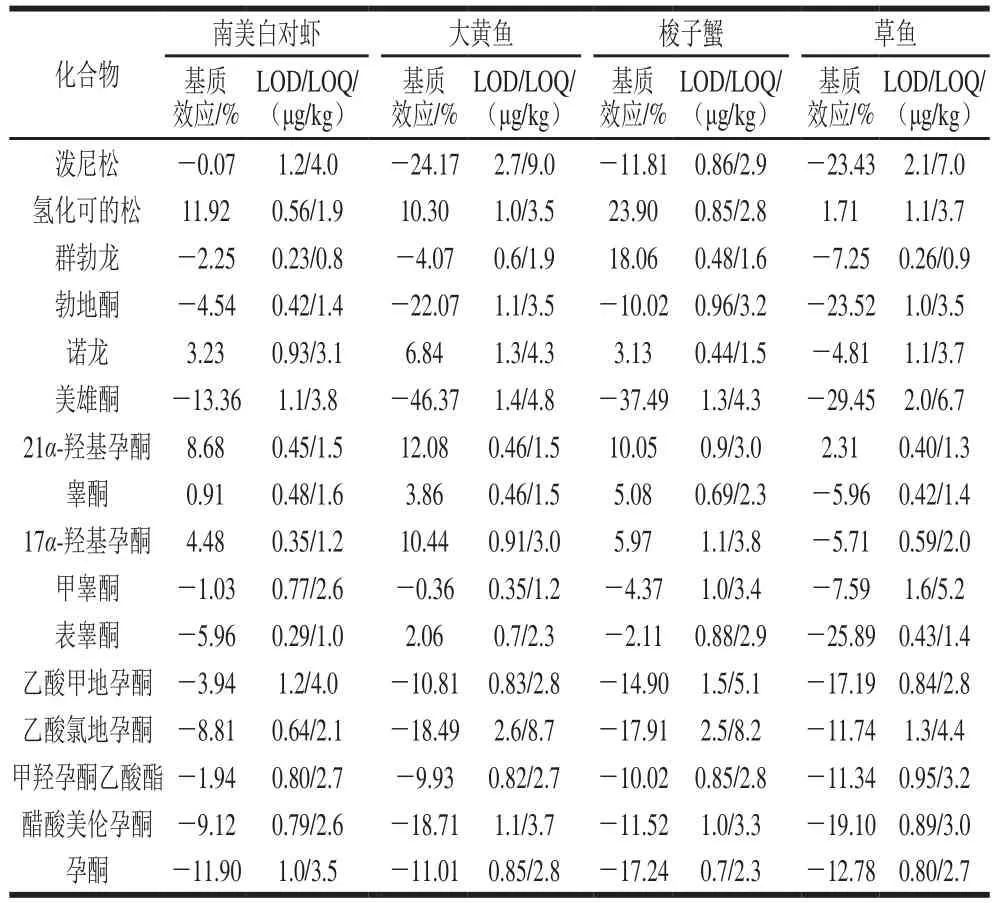
表2 激素的基质效应、方法LOD和LOQTable 2 Matrix effects, LODs and LOQs for all matrices tested
将4 种空白基质样品按1.3.2节处理后,添加不同质量浓度(1、5、10、25、50、100 μg/L)的混合标准溶液,以各激素母离子的峰面积与同位素内标的峰面积的比值(Y)为纵坐标,基质标准溶液的质量浓度(X)为横坐标,绘制标准曲线。各激素在1~100 μg/L范围内线性关系良好,16 种激素在南美白对虾、大黄鱼、梭子蟹、草鱼4 种基质中的基质匹配标准曲线的相关系数(r2)分别为0.994 5~0.999 9、0.993 8~0.999 9、0.990 6~0.999 9、0.996 4~0.999 6。
以3 倍信噪比为检出限(limit of detection,LOD),10 倍信噪比为定量限(limit of quantitation,LOQ)。如表2所示,16 种激素在南美白对虾基质中的方法LOD为0.2~1.2 μg/kg,LOQ为0.8~4.0 μg/kg;在大黄鱼基质中的方法LOD为0.4~2.7 μg/kg,LOQ为0.8~4.0 μg/kg;在梭子蟹基质中的方法LOD为0.4~2.5 μg/kg,LOQ为1.5~8.2 μg/kg;在草鱼基质中的方法L O D为0.3~2.1 μ g/k g,LOQ为0.9~7.0 μg/kg。
2.3.3 回收率和精密度
在10、20、50 μg/kg三个添加水平下做回收率实验,以考察方法的准确性和精密度(表3),16 种激素在南美白对虾基质中的回收率为64.1%~102.8%,相对标准偏差(relative standard deviation,RSD)为0.9 3%~7.6%;在大黄鱼基质中的回收率为65.2%~98.6%,RSD为1.9%~10.9%;在草鱼基质中的回收率为68.8%~114.4%,RSD为1.2%~9.2%;在梭子蟹基质中,除孕酮和泼尼松外,其余14 种激素的回收率为60.5%~121.0%,RSD为1.6%~10.4%,孕酮和泼尼松的回收率分别为53.6%~60.8%和91.7%~135.2%,RSD分别为2.3%~4.5%和2.7%~7.6%,这可能是由于梭子蟹基质中存在一些无法去除的杂质造成的基质抑制或基质增强效应,但本方法仍可保证二者的回收率处于相对稳定的状态。总体上,本研究建立的方法可满足水产品中16 种激素的实际检测要求。
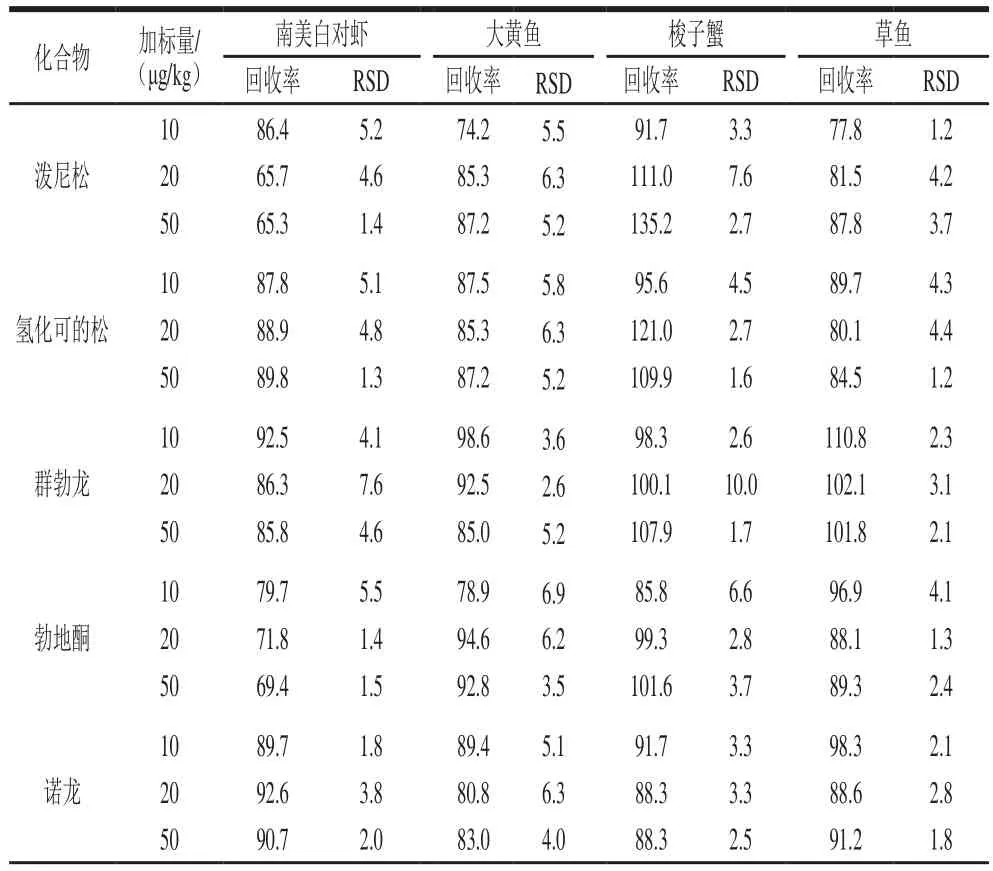
表3 激素类化合物的回收率和相对标准偏差(n=6)Table 3 Recovery and repeatability (RSD) for all matrices tested (n=6)%
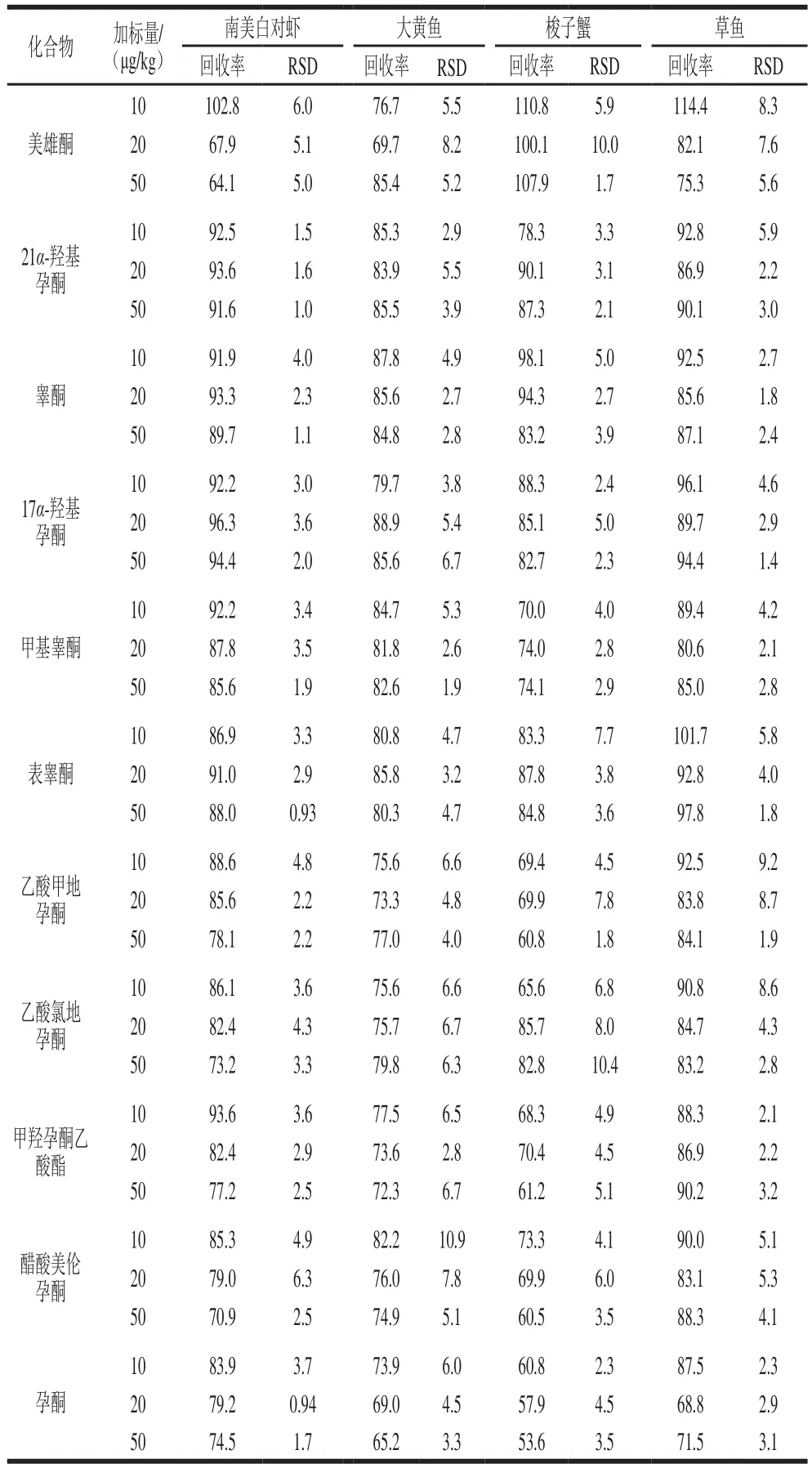
续表3
2.4 实际样品检测结果
采用本方法对36 份水产品样品(南美白对虾、大黄鱼、草鱼、鲤鱼)进行检测,其中大黄鱼和南美白对虾样品均无检出,6 份草鱼样品检出氢化可的松,含量为5.9~204.1 μg/kg,8 份鲤鱼样品中检出氢化可的松,含量为12.3~105.6 μg/kg。氢化可的松是天然存在的一种肾上腺糖皮质激素,同时也可由人工合成使用。孙利东等[31]对鸡肉和牛奶中氢化可的松的本底值进行测定,其结果在0.06~0.43 μg/kg范围内,这表明生物体内氢化可的松的本底值较低。但本次实际检测样品中检出的氢化可的松含量相对较高,外用导致的可能性较大。这与李晴[32]、蔡惠坚[33]等分别在宝石斑鱼、草鱼中检出氢化可的松的报道一致,表明在此类水产品中存在使用该激素的可能。目前我国农业部235号公告[8]规定允许在动物源食品中使用氢化可的松,且无最高残留限量要求,而文献[34]规定了牛奶中氢化可的松的最大残留限量为10 μg/kg,表明氢化可的松仍有危害人体健康的可能,因此针对水产品中氢化可的松的残留,也应提前开展相关的风险监测工作。
3 结 论
本实验优化了QuEChERS前处理方法,并结合UPLCQ-TOF MS技术,建立快速筛查鱼、虾、蟹等水产品中16 种激素残留的方法。本方法前处理简单高效,目标激素回收率满足筛查需求,同时结合高分辨质谱,使筛查结果更加准确可靠,是开展水产品中多激素残留快速筛查的一种有效手段。
