Hemocytes transcriptome pro file of the Chinese mitten crab(Eriocheir sinensis)
2018-10-17XiowuChenYweiShenYnwuiBiXugnWu
Xiowu Chen,Ywei Shen,Ynwui Bi,Xugn Wu,b,c,∗
aNational Demonstration Center for Experimental Fisheries Science Education,Shanghai Ocean University,Shanghai 201306,China
bShanghai Engineering Research Center of Aquaculture,Shanghai Ocean University,Shanghai 201306,China
cShanghai Collaborative Innovation for Aquatic Animal Genetics and Breeding,Shanghai 201306,China
Keywords:Eriocheir sinensis Hemocytes Transcriptome
ABSTRACT The Chinese mitten crab(Eriocheir sinensis)possesses an open circulatory system,in which hemolymph moves through interconnected sinuses or spaces surrounding organs called hemocoels.The hemocytes are classi fied into hyalinocytes,semigranulocytes,and granulocytes;and limited transcriptomics research is available.In this study,the transcriptome of the crab E.sinensis hemocytes was characterized.A total of 14,380,229 clean reads representing a total of 4.31 Gb nucleotides dataset were produced.A total of 67,047 contigs were obtained and 12.49%(8375)and 9.74%(6533)of the contigs were matched to data publicly available from the GenBank nr nucleotide and Uniprot databases,respectively.Among these contigs,4344 contigs belong to three categories of Gene Ontology,126 contigs to 21 subcategories of KEGG,and 4962 contigs to 25 categories of the COG database.A total of 508,379 and 345 transcripts of the E.sinensis showed>40%sequence similarity with transcripts expressed in the vertebrate blood cells from tilapia(Oreochromis niloticus),Chinese softshell turtle(Trionyx sinensis)and chicken(Gallus gallus)respectively.A total of 53,077 single nucleotide polymorphisms were identi fied and 9912 SSRs for microsatellite mining were found.The most frequent repeat motifs detected were dinucleotide repeats,accounting for 37.52%of the total repeats.Our data provides an important gene resource for the exploitation of molecular marker-assisted breeding,immune mechanisms and conservation of germplasm in the crab E.sinensis.
1.Introduction
The Chinese mitten crab(E.sinensis)has been widely introduced into many regions outside of its native habitat in China where it has long been an economically important aquaculture species and a traditional food resource.Rapid development of large-scale and highly intensive mitten crab farming raised disease outbreaks that seriously affected aquaculture production(Wang et al.,2011;Zhang and Bonami,2007).The innate immune system plays a key role in crab defense and the immune response is activated with the recognition of speci fic surface molecules unique to the different pathogens to stimulate cellular or humoral effectors(Vazquez,Alpuche,Maldonado,Agundis,Pereyra-Morales,Zenteno,2009).In E.sinensis three categories of hemocytes have been identi fied,namely,the hyaline,semigranular and granular cells.Some hemocytes contribute to phagocytosis,whereas hemocytes migration and lysis will lead to pathogen elimination(Lv et al.,2014).Many important immune-related genes from the complement and coagulation cascades,the VEGF signaling pathway,the Wnt signaling pathway,natural killer cell-mediated cytotoxicity,the MAPK signaling pathway,neuroactive ligand-receptor interactions,and the Lysosome pathway are involved in the response of the Chinese mitten crab to infection with Spiroplasma eriocheiri(Wu,Wang,Wang,Zhao,Hu,Chen,2017).
Recently,several microsatellite markers have been identi fied in genes associated with metabolism,growth,and immunity(Chang,Liang,Ma,He,&Sun,2008;Li,Geng,Chen,&Sun,2016;Xiong,Wang,Qiu,2012).Sequence tag expression analysis using first-generation sequencing technologies has been performed on hemocytes of E.sinensis(Zhao,Song,Wang,Zhang,Hu,Chen,2009).The result showed that 3118 clones were sequenced and analyzed by homology searches in the GenBank,KEGG and Uniprot.Signi ficant homology to known genes was found in 488 of the 1039 unique sequences.Therefore,the amount of data is inadequate to reflect the function of hemocytes and the relative expression level of genes.
In this study,the Illumina Hiseq2500 platform was used to characterize the transcriptome of E.sinensis hemocytes and to identify novel molecular markers involved in crab innate immunity.Additionally,we have also analyzed SSR markers as well as small variants(SNPs).This will appreciably promote further research into the immune defense mechanisms of E.sinensis.
2.Materials and methods
2.1.Sample collection and RNA extraction
Five 10-month-old E.sinensis individuals were collected from the Chongming aquafarm in Shanghai Ocean University(31°19′22″N;121°45′52″E).Approximately 500 μL hemolymph samples were extracted from the third pereiopod of each crab,diluted with the same amount of anticoagulant solution(510 mmol/L NaCl;100mmol/L glucose;200mmol/L citric acid;30mmol/L sodium citrate;and 10 mmol/L EDTA-Na2;pH 7.3),followed by centrifugation at 800 r/min,10min,4°C.The supernatant was discarded and the cell pellet was solubilized in TRIzol(Invitrogen,USA)and Total RNA was extracted according to the manufactures protocol.RNA quantity and quality were ascertained through an Agilent Technologies 2100 Bioanalyzer(Agilent Technologies,CA,USA).Genomic DNA was digested with DNase I(TaKaRa,Japan)and poly(A)mRNAs were puri fied for cDNA library construction using an Ultra™RNA Library Prep kit(NEB,China).
2.2.Constructions and sequencing of cDNA library
Illumina HiSeq library was constructed following the manufacturer's directions(Illumina,USA).Using a PCR with random-hexamer primers followed by RNAseH treatment and polymerization using Pol I,mRNA was reverse transcribed into cDNA.Production of paired-end library was performed using the Genomic Sample Prep kit(Illumina,USA).cDNA with appropriate lengths were puri fied using the QIAquick Polymerase Chain Reaction puri fication kit(Qiagen,Hilden,Germany),end repaired and tethered with sequencing adapters(Margulies et al.,2005).AMPureXP beads(NEB,USA)were utilized to exclude unbe fitting DNA pieces,and the sequence library was prepared by PCR.After veri fication by fluorospectrophotometry using PicoGreen and quanti fication by Agilent 2100 Bioanalyzer,the libraries of multiplexed DNA were mixed in equi-molar amounts to a normalized concentration of 10 nmol/L and sequenced through an Illumina HiSeq™2000 platform employing the paired-end strategy.
2.3.Data filtration,de novo assembly and bioinformatics analysis
Initial sequencing reads have been combined to perform a stringent filtration procedure and ensuing de novo assembly.After removing adapter contamination,reads were inspected in the 3′to 5’direction and in 5 bp windows in order to eliminate bases whose quality score were q<20.Furthermore,reads shorter than 25 bp in length were also excluded.
The trinity software was used to compile all clean reads,followed prediction of protein-coding regions through the GetORF module from European Molecular Biology Open Software(Itaya,Oshita,Arakawa,&Tomita,2013).All protein-coding sequences were submitted to basic local alignment search tool(BLAST)similarity searches against the National Center for Biotechnology,the Information nonredundant protein database and the EuKaryotic Orthology Groups database and hits with an E value less than 1 e-5were used to identify orthologies.The GoPipe software was used for BLASTP(cutoff E value of<1 e-5)against the Swiss-Prot and EMBL databases.GO annotation according to the categories “molecular function”“biological process”and “cell component”was performed using the gene2go software(Ashburner et al.,2000).Likewise,the deduced protein sequences were subjected to BLASTP(cutoff E value of<1 e-3)using the Kyoto Encyclopedia of Genes and Genomes(KEGG)database to allocate KEGG Orthology(KO)numbers.Pathways related to metabolism were predicted on the basis of the KO assignment,with KEGG tools(Kanehisa,Goto,Furumichi,Tanabe,&Hirakawa,2010).The mapped read number of each gene was determined by the number of bp per read and sequencing depth;whereas the reads per kb per million reads(RPKM)were estimated for gene expression level normalization(Mortazavi,Williams,McCue,Schaeffer,Wold,2008).
2.4.SSR and SNP mining
PerlscriptMISA (http://pgrc.ipk-gatersleben.de/misa/;Thiel,Michalek,Varshney,Graner,2003)was employed to recognize SSRs.In SSR analysis,the smallest number of nucleotide repeats was 8 for dinucleotides,6 for tri-nucleotides and 5 for tetra-,penta-and hexa-nucleotides,respectively.For complex SSRs,the maximum interval between two sets of repeats was allowed for 50 bp.SNPs in all unigenes were searched and generated using Samtools(http://samtools.sourceforge.net)and Varscan(http://varscan.sourceforge.net).The total number of SNPs were calculated across the database.
2.5.Comparative expression analysis across species
RPKM using the DEGseq package based on the MA-plot-based method using random sampling model was directly utilized to compare gene expression levels(Wang,Feng,Wang,Wang,Zhang,2010).The Benjamini-Hochberg technique was used to establish the value that would be the most appropriate signi ficance cut-offin several tests.In this work,“q < 10-3”and “|log2(RPKM a/RPKM b)|≥2”were selected to recognize species-biased genes.Expression data from the blood cells of Oreochromis niloticus,Trionyx sinensis and Gallus gallus come from our company work.
3.Results
3.1.Transcriptome sequencing and assembly
A total of 14,380,229(98.93%)clean reads were obtained from 14,535,254 raw reads after filtering adaptor and short sequences(<40 bp),as well as low-quality and “N”bases using FASTX-Toolkit(http://hannonlab.cshl.edu/fastx_toolkit/).Total reads were compiled into 67,047 contigs ranging from 201–16,991 bp,with a N50 length of 542 bp(Table S1).Length statistics of the assembled contigs of E.sinensis hemocytes are described in Fig.S1.
3.2.Gene annotation
After alignment against sequences in public databases,the study generates annotation information of predicted proteins(Supplementary 1).A total of 8375 and 6533 non-redundant contigs were obtained by submitting to the GenBank Nr nucleotide and Swiss-Prot database,respectively and most corresponded to insect entries (Fig.1,Supplementary 2).
Among 67,047 transcripts,4344 were classi fied under the GO term 3 categories and subclassi fied within 49 subcategories:biological process(12,979,43.06%),molecular function(9,750,32.35%)and cellular component(7,412,24.59%)(Fig.2).A total of 126 transcripts were assigned to five pathways,namely,metabolic,cellular processes,organism system,genetic and environmental data processing with further 21 subcategories.A total of 4962 unigenes were classi fied into 25 eggNOG categories.The categories “General function prediction only”(666,13.42%)and “Signal transduction mechanism”(486,11.07%)were the two largest functional groups(Fig.3).
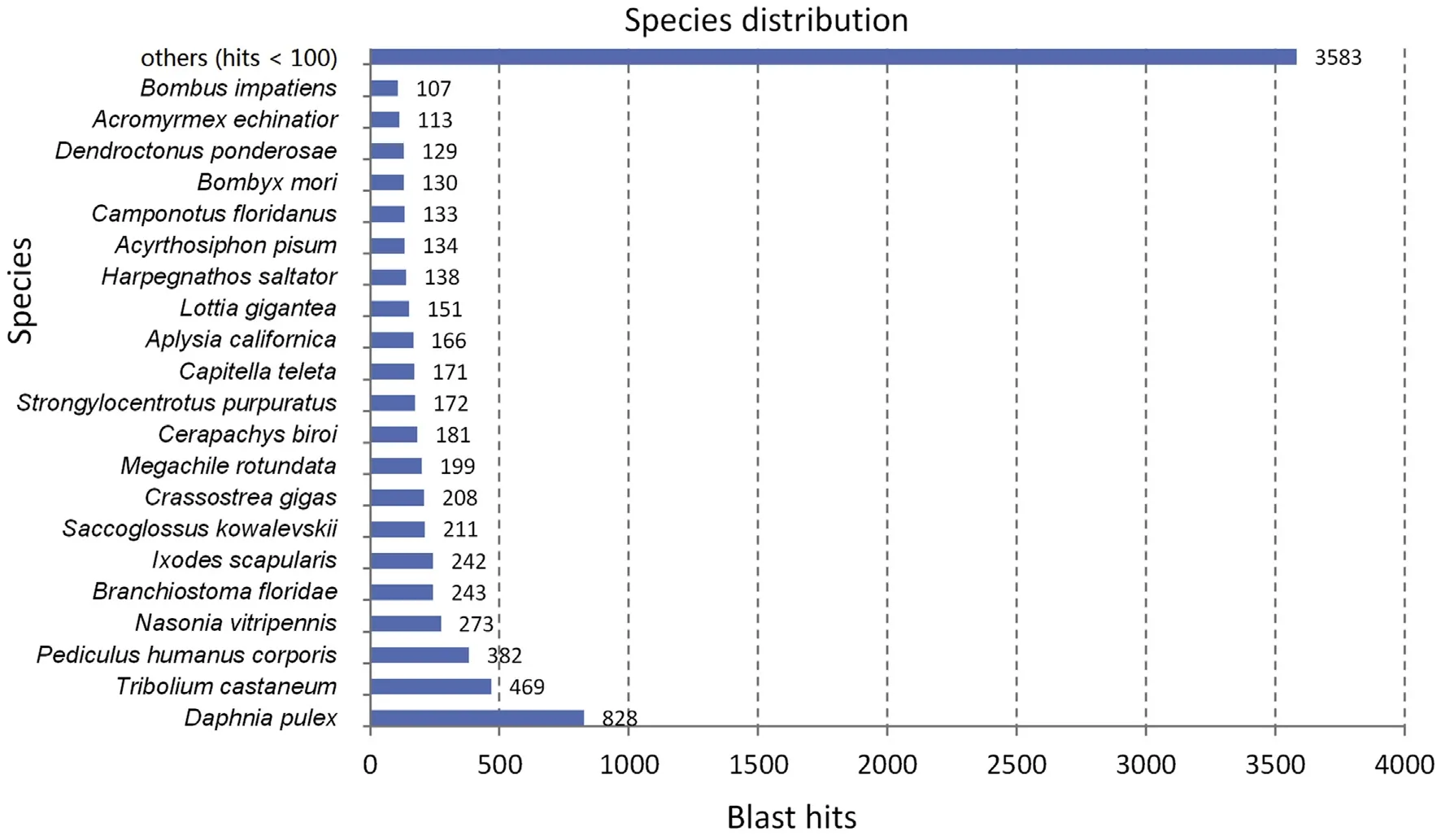
Fig.1.Distribution of the BLAST hits in function of the species.
3.3.SSRs and SNPs identi fication
In total,we identi fied 9112 SSRs,distributed in 8334 sequences.The frequency distribution for those SSRs with mono-,di-,tri-,tetra-,and penta-nucleotide repeats were 25.28%,37.52%,28.09%,1.96%,0.03%,respectively.A total of 53,077 SNPs were identi fied in 10,065 sequences.In 53,077 SNPs,heterozygotes account for 99.87%,homozygotes number account for 0.13%,transition number account for 67.89%,and transversion number account for 32.11%.The SNP conversion of G/A(9,992,18.83%),T/C(8,102,15.26%),C/T(9,588,18.06%),A/G(8,357,15.75%)are the most abundant(Fig.S2).The raw sequence data from E.sinensis hemocyte was uploaded in the Sequence Read Archive database(Accession numbers:SRP059617).
3.4.Comparison to vertebrates
The transcripts present in E.sinensis hemocytes were compared with the blood cells transcriptome from the vertebrates tilapia(Oreochromis niloticus),Chinese softshell turtle(Trionyx sinensis)and chicken(Gallus gallus)(from our accompanying work).A total of 508,379 and 345 transcripts of E.sinensis showed>40%sequence similarity with O.niloticus,T.sinensis and G.gallus(Supplementary 3).Overall,there are 746 crab transcripts in at least one of the three vertebrate groups and 152 transcripts are present cross all the groups(Fig.4).

Fig.2.Functional annotation of assembled sequences based on gene ontology(GO)categorization.

Fig.3.Eukaryotic of orthologous groups(KOG)classi fication of assembled contigs.
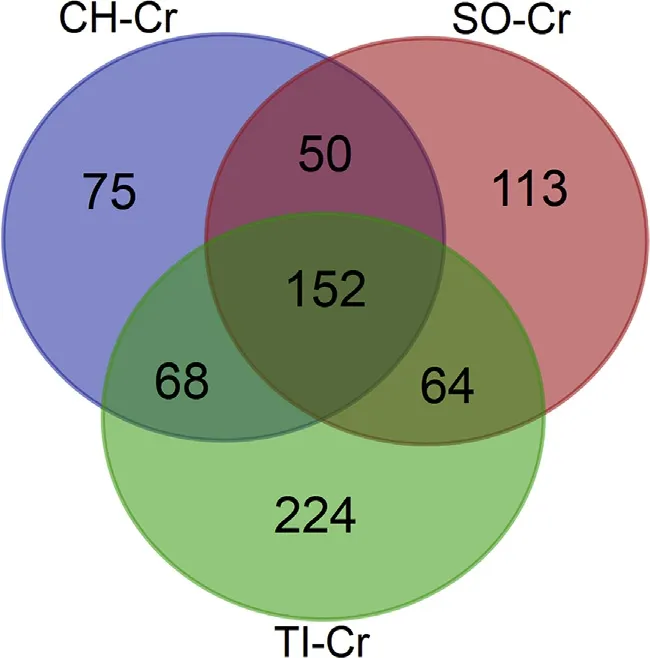
Fig.4.List of the most similar transcripts between crab and vertebrates.Purple,red and green circles represent the three groups of G.gallus vs.E.sinensis,T.sinensis vs.E.sinensis and O.niloticus vs.E.sinensis groups,respectively.(For interpretation of the references to colour in this figure legend,the reader is referred to the Web version of this article.)
4.Discussion
High throughput transcriptome sequencing has been extensively employed to characterize the gene expression pattern,detection of alternative splice variants and mutations in the tissues of several species(Morozova,Hirst,Marra,2009).SSRs are tandemly repeated sequences of one to six nucleotides which occur abundantly in all eukaryotic and prokaryotic genomes(Field&Wills,1996;Weber,1990).They are uniformly distributed throughout the genomes and frequently showed high level of length polymorphism and are a very informative and a useful tool to characterize genomic variation for gene linkage analysis and association studies(Gulcher,2012;Vieira,Santini,Diniz,Munhoz Cde,2016).SNP refers to a change in the genome DNA sequence that results from a single nucleotide change that can be caused by a single base transition or transversion or by insertions or deletions.SNPs are responsible for the occurrence of various human diseases such as tumor,infectious diseases,autoimmune diseases,neuropsychiatric disorders,sickle-cell anemia and cystic fibrosis(Syvanen,2001).Conventionally,the identi fication of SSR markers depended on screening genomic DNA libraries through continuous probes,followed by the positive clonesequencing for developing SSR primers(Zane,Bargelloni,Patarnello,2002).Nevertheless,the majority of such experiments are time-consuming and ineffective.Next-generation sequencing techniques could promote the efficiency of generating a large number of DNA sequences useful for isolating SSR markers from non-model animals.
The immune responses of crustaceans rely on their innate immunity,which is activated after recognition of pathogen-related molecular patterns by various host proteins,including lectin,anti-microbial proteins,pattern recognition proteins and clotting.These proteins subsequently activate cellular or humoral effector systems that will kill the foreign pathogens(Vazquez et al.,2009).The E.sinensis possesses an open circulatory system,in which hemolymph moves through interconnected sinuses or spaces surrounding organs called hemocoels.Till now,only two transcriptomics research have been performed to analyze the gene expression of hemocytes from the E.sinensis,while the sequencing data is not large(Wu,Wang,Wang,Zhao,Hu,Chen,2017;Zhao,Song,Wang,Zhang,Hu,Chen,2009).A small proportion of hemocytes contribute to the phagocytosis,and hemocytes migration and lysis contribute a major pathway for eliminating pathogens(Lv et al.,2014).There have been reports on the role of hemocytes in the immune system of the E.sinensis.The mRNA expression levels of the suppressor of cytokine signaling in hemocytes were notably up-regulated after the activation with polyinosinic-polycytidylic acid,lipopolysaccharide and Aeromonas hydrophila,which demonstrated that EsSOCS6 might regulate the activation of the NF-κB signaling pathway and play a signi ficant role in the innate immune responses of E.sinensis(Qu et al.,2018).Expression of Receptor for activated C kinase 1(RACK1)mRNA in hemocytes was considerably up-regulated following the activation with Pichia pastoris and Vibrio anguillarum,which indicated the role of RACK1 in the innate immunity of E.sinensis(Jia,Wang,Wang,Wang,&Song,2016).A prophenoloxidase(proPO)gene was cloned from haemocytes of E.sinensis;the mRNA expression pattern and activity fluctuation of proPO stimulated by Vibrio anguillarum showed that it may be essential for the quick action against pathogens in E.sinensis(Gai et al.,2008).Although there are some immune genes reported in E.sinensis,that is still very lacking of related information.The biggest challenge of function analysis of crab genes is that there is no reference model organism,so it is difficult to de fine or classify the gene name.In this study,using high-throughput sequencing,we have identi fied 67,047 unigenes in total.The majority of the assembled unigenes(87.50%)did not match to any nucleotide or protein available from the public database.Thus,data produced from this study largely enriched the transcriptome data of E.sinensis.In addition,746 genes showed high similarity to three vertebrates,that are will be the key emphasis of study in the future.
Gene homologues of the vertebrate ILF2,TGF-β,major histo-compatibility complex(MHC)were identi fied in E.sinensis.Fish and crab share more high similarity genes than compared to other vertebrate groups.Animal immune systems evolved earlier in evolution and some immune blocks were moderated,a few obtained different function while others were lost.Invertebrates contain cellular receptors,which attach to pathogens and differentiate non-self from self.Cytokines,such as ILF2,TGF-β,participate in this immune response which ultimately eliminate or inactivate the intruder.Fish homologous cytokine related signal molecules can also be found among invertebrates.The cartilaginous and bony fishes,amphibians,reptiles,birds and mammals developed the MHC,B-cell and T-cell receptors as supplementary adaptive processes to support innate immunity(Buchmann,2014).The MHC,as a key element in adaptive immunity,is present in fish but its existence in invertebrates remains unclear.A common ancestral locus found in the early chordates is named as the proto-MHC,which might be the initial prototype of the MHC.It was possibly established in evolution through chromosome duplications(Kasahara,Suzuki,&Pasquier,2004).But,relatively speaking,the E.sinensis showed very low similarity of the gene sequences to vertebrates.More information is needed to study the gene categories and functions of invertebrates.Such as the identi fication of protein domains which may provide more relevant information of unannotated gene function.
The tandem repeat LEI0258 is a genetic marker which is located within the B locus of chicken MHC and it is involved in humoral and cellular immune responses(Esmailnejad,Nikbakht Brujeni,&Badavam,2017).The cytokine Macrophage Migration Inhibitory Factor(MIF)is an immunoregulatory factor encoded in a functionally polymorphic locus thus linking the gene to autoimmune and infectious diseases.The MIF gene promoter consists of a 4-nucleotide microsatellite polymorphism.Also,the UHRF1 transcription factor is the main MIF-interacting partner.UHRF1 is essential for MIF transcription(Yao et al.,2016).
We performed high-throughput sequencing of hemocytes from the E.sinensis.The data provide a useful basis to investigate ecologically pertinent genetic variations of the Chinese mitten crab and also provides more information about the acquisition of primary information of immune-related genes,which may subsequently be utilized to comprehend microsatellite-related immune genes.
Acknowledgments
This research was supported by General Project from the Natural Science Foundation of China(No.31572630),The capacity promoting project of Shanghai Engineering and Technology Center from Shanghai Municipal Science and Technology Commission(No.16DZ2281200)and Shanghai Universities Top Disciplines Project of Fisheries(No.2015-0908)from Shanghai Education Commission.
Appendix A.Supplementary data
Supplementary data related to this article can be found at https://doi.org/10.1016/j.aaf.2018.07.006.
杂志排行

Aquaculture and Fisheries的其它文章
- Polymorphisms in the FOXO gene are associated with growth traits in the Sanmen breeding population of the razor clam Sinonovacula constricta
- GCRV 096 VP6 protein and its impacts on GCRV replication with different genotypes in CIK cells
- Cloning and expression analysis of the nuclear factor erythroid 2-related factor 2(Nrf2)gene of grass carp(Ctenopharyngodon idellus)and the dietary effect of Eucommia ulmoides on gene expression
- Positive impacts of Nile tilapia and predatory sahar on carp polyculture production and pro fits
- Assessment of post-harvest fish losses Croaker Pseudotolithus elongatus,(Bowdich,1825),Cat fish Arius heudeloti,(Valenciennes,1840)and Shrimp Nematopalaemon hastatus(Aurivillius,1898)in Ondo State,Nigeria