柱前衍生/高效液相色谱法测定烟用添加剂中的咪唑
2018-10-16程利侠何东香杨金龙吕长平
程利侠,何东香,杨金龙,吕长平
(甘肃烟草工业有限责任公司技术研发中心,甘肃 兰州 730050)

图1 丹磺酰氯与仲胺类的反应式Fig.1 Reaction of Dns-Cl and secondary amine
烟用添加剂是卷烟产品的重要组成部分,可增强或改善烟草产品的香味或吃味,以满足消费者的需求[1-2]。但在烟用添加剂生产过程中,可能引入微量的有害成分,如邻苯二甲酸酯类、工业甲醇、咪唑等化合物。其中,咪唑(Imidazole)是一种氮杂环化合物,又称1,3-二氮唑,为白色结晶固体,易溶于水及乙醇;易与酸生成盐,遇碱易脱去氢;分子间存在氢键缔合,沸点较高(256 ℃)。咪唑分子中存在一个6电子共轭大π键,故具有典型的芳香性[3],工业上可由乙二醛、甲醛与氨反应或用咪唑-4,5-二羧酸经脱羧制得[4]。
目前,文献报道的甲基咪唑化合物(2-or 4-甲基咪唑)的检测方法主要有紫外光谱法[5]、气相色谱法[6-9]和液相色谱法[10-14],而对咪唑的检测方法报道甚少。目前烟草行业使用的烟用添加剂种类繁多,成分复杂[15],且咪唑的沸点高,采用气相色谱法分析烟用添加剂中的咪唑时,存在定性较困难、准确度较低且在色谱柱中残留严重等问题。另外,咪唑是小分子化合物,直接测定时不易分离,因此需先衍生化以引入荧光和紫外发光基团,提高其分离度,以易于色谱分析。
丹磺酰氯(Dansyl chloride,Dns-Cl)是一种常用的柱前衍生化试剂,被广泛用于生物胺[16]、组胺[17]和咪唑类[11]化合物的柱前衍生化,具有很好的专一性,可定量完成磺酰化反应,操作简单,衍生物稳定性好。其反应原理如图1所示:在一定条件下,丹磺酰氯可与仲胺基上的活泼氢反应,脱掉1分子的HCl生成具有荧光和紫外吸收的衍生物[16]。
由于咪唑在结构上具有仲胺基活泼氢,本实验先在碱性条件下用丹磺酰氯作为柱前衍生化试剂,将咪唑转化成易于检测的具有荧光和紫外吸收的衍生物,再经高效液相色谱/荧光检测器分离分析。方法灵敏度高,基体干扰小,可获得理想的实验结果。
1 实验部分
1.1 仪器、试剂与材料
Agilent 1200 型高效液相色谱仪,荧光检测器(美国Agilent公司);色谱柱为XBridgeTMPhenyl C18柱(4.6 mm×250 mm×5 μm,美国Waters 公司);预柱为Acclaim®Explosive E2 C18柱(10 mm×4.3 mm,5 μm,美国Dionex公司)。ATL-224-1精密电子天平(德国赛多利斯公司,感量0.1 mg);Millipore超纯水发生器;AnkeTGL-10B高速台式离心机(上海安亭科学仪器厂);KQ-500DE超声波清洗仪(昆山市超声波清洗仪器有限公司);LABORTA4003旋转蒸发仪(德国海道夫公司);CWF1100马弗炉(英国Carbolite Furnawws公司);30 mL离心管(PP材质,Corning公司);有机相滤膜(0.45 μm);TM-1旋涡混合器(无锡沃信仪器有限公司)。
烟用添加剂:从不同生产厂家获取的烟用香精香料。丙酮(分析纯及以上,天津市光复精细化工有限公司);乙腈、乙酸乙酯(色谱纯,美国Tedia公司);丹磺酰氯(纯度>99.5%,上海TCI公司);咪唑标准品(纯度>99.5%,北京百灵威公司);咪唑衍生物(自制,咪唑衍生化后经层析柱分离得到);无水硫酸钠(分析纯,天津市光复精细化工有限公司)于150 ℃烘至2 h,冷却后置于干燥器中备用;实验用水为美国Millipore纯水系统制备的去离子水。
1.2 标准溶液的配制
1.2.1咪唑标准储备液称取500 mg咪唑用水溶解后定容至100 mL容量瓶中,配成5 g/L的储备液,储存于0~4 ℃,有效期1个月。根据需要配制合适浓度的系列标准工作溶液。
1.2.2标准工作溶液以水作溶剂,将咪唑标准储备液配制成0.5、1.0、5.0、10.0、50.0、100.0、500.0 mg/L系列工作标准溶液,贮存于0~4 ℃,有效期7 d。
1.2.3衍生化试剂配制称取1.0 g丹磺酰氯用丙酮溶解并定容至100 mL棕色容量瓶中,质量浓度为10 g/L,即用即配。
1.2.4衍生化系列工作溶液分别取咪唑系列标准工作溶液1.0 mL于离心管中,用4.0 mL衍生化试剂进行衍生化反应,然后根据样品处理方法萃取浓缩标准溶液。用衍生化标准工作溶液制备标准曲线,以保留时间定性,根据咪唑标准工作溶液的浓度及其衍生物响应峰面积,外标法定量。
1.3 样品处理
1.3.1烟用添加剂样品的处理称取约5.0 g样品于离心管或锥形瓶中,加入2 mL pH 10.0的Na2CO3-NaHCO3缓冲溶液,涡旋并摇匀后放置10 min,再加入4.0 mL衍生化试剂,于45 ℃下超声振荡衍生化40 min,反应结束后取出离心管,再加入1 mL 0.5 mol/L盐酸溶液并摇匀终止反应。再用40 mL乙酸乙酯分两次萃取反应液,分出有机层,合并有机层至50 mL锥形瓶中,并用适量的Na2SO4干燥过夜,干燥后的样品过滤至50 mL浓缩瓶中,滤液用旋转蒸发仪于50 ℃蒸至0.2 mL左右,然后用乙腈定容至1 mL,并过0.45 μm有机滤膜,滤液待测。对于难蒸干的样品,将旋蒸残余的液体用少许乙腈清洗,并全部转移至微量器中,过0.45 μm有机滤膜,滤液待测。对于萃取不分层的样品,将溶液转移至离心管中,以4 000 r/min离心5 min,然后吸取上层清液并用无水Na2SO4干燥过夜,干燥后的样品过滤至50 mL浓缩瓶中,滤液用旋转蒸发仪于50 ℃蒸至0.2 mL左右,然后用乙腈定容至1 mL,并过0.45 μm有机滤膜,滤液待测。
1.3.2空白样品的制备于干净离心管或锥形瓶中(不加香精香料或咪唑标准品),加入2 mL pH 10.0的Na2CO3-NaHCO3缓冲溶液,涡旋并摇匀后放置10 min,加入4 mL衍生化试剂,然后于45 ℃下超声振荡衍生化40 min,反应结束后取出离心管,再加入1 mL 0.5 mol/L盐酸溶液并摇匀终止反应。最后用乙酸乙酯萃取、浓缩、乙腈定容至1 mL,并过0.45 μm有机滤膜,滤液待高效液相色谱分析。
1.4 液相色谱条件
柱温:30 ℃;流速:1.0 mL/min;进样量:20 μL;流动相:A(水),B(乙腈),梯度洗脱程序:0~2 min,30%B;2~10 min,30%~70%B;10~25 min,70%B;25~26 min,70%~30%B;26~31 min,30%B。检测波长:335 nm(λex:335 nm,λem:503 nm)。
若待测试样溶液的浓度超出标准曲线的浓度范围,则对样品前处理适当调整后重新测定。
2 结果与讨论
2.1 萃取条件的选择
由于烟用添加剂样品复杂,有些既难溶于水,也难溶于有机溶剂。为完全萃取样品中的待测成分,本实验选用乙酸乙酯分批萃取衍生化后的烟用添加剂样品,并考察了萃取体积对咪唑含量测定的影响,结果发现,当萃取体积为30 mL时,测定值达到最大,继续增加萃取体积,其值基本稳定。为保证萃取充分,实验选择萃取体积为40 mL。
2.2 衍生化条件
以咪唑标准品为样品,丹磺酰氯为衍生剂,研究了Na2CO3-NaHCO3缓冲溶液的pH值、衍生化时间、衍生化温度、衍生剂用量对衍生反应的影响。
2.2.1缓冲溶液pH值的影响咪唑在碱性条件下易失去质子,因此研究反应体系的pH值至关重要。本实验考察了Na2CO3-NaHCO3缓冲溶液在不同pH值(8.5、9.0、9.5、10.0、10.5、11.0)下对衍生产物峰面积的影响。结果显示pH 10.0时衍生化产物的生成量最大,因此本文选取缓冲溶液体系的pH值为10.0。
2.2.2衍生化反应时间的影响取相同浓度(10.0 mg/L)的咪唑溶液6份,分别加入过量的丹磺酰氯,超声振荡不同时间(10、20、40、60、80、100 min),以考察衍生化反应时间的影响。结果表明,反应40 min时,衍生产物的生成量最大,因此选择衍生时间为40 min。
2.2.3衍生温度的影响在固定咪唑和衍生剂用量的条件下,考察了衍生温度分别为15、25、35、45、55、65 ℃时衍生产物的生成量。结果表明衍生温度为45 ℃时,衍生产物量最多,此后随衍生温度的继续增加,衍生物产量不变,因此选择衍生化温度为45 ℃。
2.2.4衍生剂(Dns-Cl)用量的影响取相同质量浓度(10.0 mg/L)的咪唑溶液6份,考察了衍生剂分别为10、50、100、500、1 000、2 000 mg/L时对咪唑衍生程度的影响。每组做3次平行实验,取3次测量的平均值。测定结果显示,衍生剂浓度过量100倍时,咪唑衍生化反应最好,衍生物的生成量最多。因此选择过量100倍(1 000 mg/L)的衍生剂用量,以保证既不会造成试剂浪费,又能反应完全。

图2 咪唑标准品衍生化产物(a)和烟用添加剂样品衍生化产物(b)的分离色谱图Fig.2 Separation chromatograms of the imidazole standard de-rivative(a) and derivative of cigarette additive sample(b)
2.3 流动相的选择
咪唑衍生产物的极性不大,在水中的溶解性较小,流动相中需含有大量的有机溶剂。常用有机溶剂有甲醇(质子性溶剂)、乙腈(非质子性溶剂),对于含氮化合物,乙腈的洗脱能力更强。实验分别采用乙腈-水、甲醇-水作为流动相,发现前者作为流动相得到的峰形比后者好,通过调节乙腈-水的体积比,梯度洗脱程序,使待测成分与样品中的其它成分得到了较好的分离。在此条件下,考察了咪唑标准品衍生化产物和烟用添加剂样品衍生化产物的液相分离色谱图(图2)。结果表明:在选定的流动相条件下,样品中的色谱峰得到很好的分离,且峰形良好。
2.4 检测波长的选择
经荧光检测器的光谱扫描,色谱图中衍生化产物的最大激发波长(λex)为335 nm,最大发射波长(λem)为503 nm,实验选择335 nm为检测波长,此时测定灵敏度较高且色谱峰分离较好。
2.5标准曲线、检出限与定量下限
按优化色谱条件对系列衍生化工作溶液进行分析,测得不同浓度标样的峰面积响应值。以峰面积响应值(y)为纵坐标,标准物质的质量浓度(x,mg/L)为横坐标进行线性回归分析。咪唑在0.5~500 mg/L范围内线性关系良好,相关系数(r)为0.999 5。将空白样品溶液重复测定10次,计算标准偏差,以3倍标准偏差计算得到方法的检出限(LOD)为0.24 mg/kg,以10倍标准偏差计算得到方法的定量下限(LOQ)为0.80 mg/kg。
2.6 回收率及相对标准偏差
选择实际样品添加0.5、10.0、500 mg/L水平的咪唑标准溶液,按上述样品处理方法和优化色谱条件进行分析,得到3个加标水平下咪唑的回收率为89.1%~97.2%,相对标准偏差(RSD)为2.8%~6.2%。在相同条件下对同一样品平行测定6次(每隔1 d测定1次),计算日间精密度,得其RSD不大于7.5%。结果表明本方法能准确测定烟用添加剂中咪唑的含量。
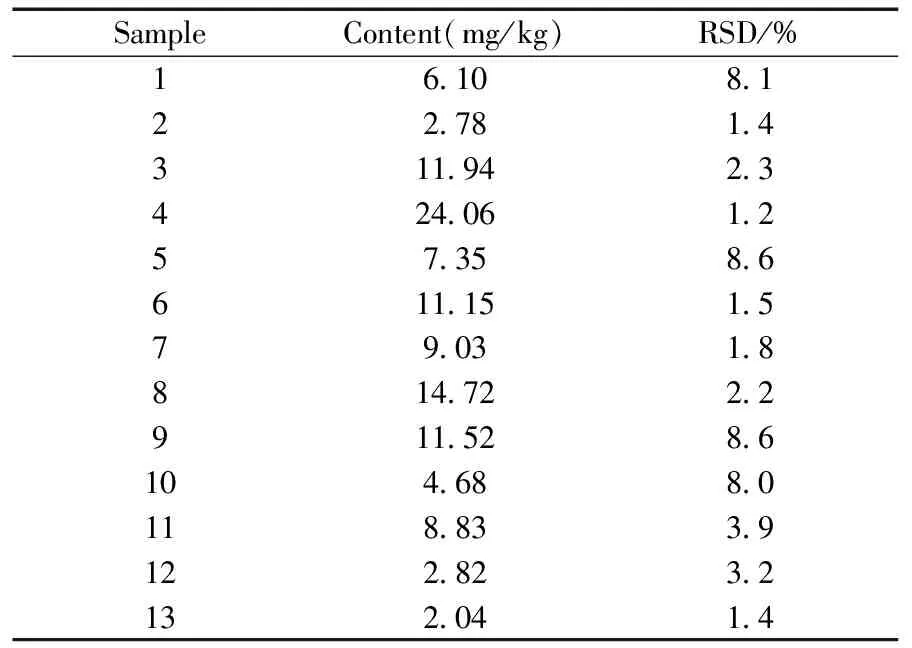
表1 不同烟用添加剂样品的测定结果Table 1 Detection result of the different cigarette additive samples
* undetected samples were not listed
2.7 实际样品的测定
在上述实验条件下,测定了60个不同种类的烟用添加剂样品中的咪唑含量。实验结果显示:仅13种烟用添加剂中检出咪唑化合物(见表1),且含量均较低(未超过25 mg/kg)。
3 结 论
本文建立了柱前衍生/高效液相色谱测定不同烟用添加剂中咪唑含量的方法,优化得到了最佳衍生化试剂条件。方法的检出限为0.24 mg/kg,定量下限为0.80 mg/kg,加标回收率为89.1%~97.2%。方法适用于烟用添加剂中咪唑含量的测定,可为保障烟用添加剂的质量安全提供依据。
