沈阳地区2013—2017年手足口病肠道病毒柯萨奇A组6型的分子流行病学研究
2018-10-15王冰张春青王萍白丽娜李天宝安向东陈叶
王冰 张春青 王萍 白丽娜 李天宝 安向东 陈叶
110031沈阳市疾病预防控制中心
肠道病毒(enterovirus,EV)是一类无包膜单股正链RNA病毒,属于小RNA病毒科肠道病毒属,人类是其最重要的宿主。EV主要通过粪-口途径感染,引起急性弛缓性麻痹、疱疹性咽峡炎、手足口病、无菌性脑膜炎和结膜炎等多种临床症状。根据遗传特征可将EV分为A、B、C和D四个病毒基因组,手足口病主要由A组的肠道病毒A71型(enterovirus A71, EV-A71)、柯萨奇病毒(coxakievirus, CV)A 组(2、4、5、6、10、16 型)和 B 组(1 ~5 型)等引起,以往监测表明EV-A71和CV-A16最为常见[1]。然而,从2013年开始,我国多个省份监测显示柯萨奇病毒A组6型(CV-A6)开始增多且引起多地爆发,成为手足口病的主要病原之一,甚至有取代EV-A71和CV-A16的趋势。
因此,为了解辽宁省沈阳地区CV-A6型病毒的分子流行特点,本实验室选取了2013—2017年间收集的手足口病非EV-A71非CV-A16肠道病毒阳性标本进行CV-A6的检测,并对其VP1全长基因序列进行测定和系统进化分析,为今后此类疾病监测和疫情处置提供客观的参考依据。
1 材料方法
1.1 标本收集 标本均取自2013—2017年辽宁省沈阳地区各级手足口病哨点监测医院收治的疑似手足口病患者的粪便、肛拭子以及咽拭子标本,共2 881 份,男女患者比例为 1.43∶1(1 696∶1 185)。 标本采集、运输和保存等操作均严格按照《手足口病预防控制指南(2009年版)》进行。
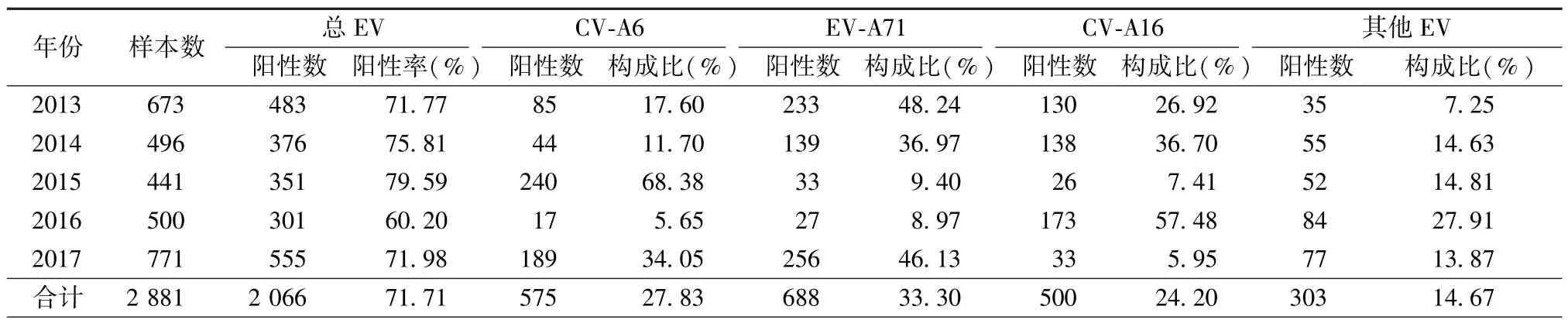
表1 沈阳地区2013—2017年手足口病病原构成及肠道病毒阳性病例数Tab.1 Enterovirus-associated hand,foot and mouth diseace cases and distribution of enterovirus strains in Shenyang from 2013 to 2017
1.2 标本前处理 取粪便标本约0.1 g,加入分装好0.9 ml PBS的1.5 ml Eppendorf管中。振荡混匀,制备10%的便悬液,5 000×g离心5 min,应用上清液进行RNA提取。肛拭子标本参考粪便标本进行前处理,咽拭子标本振荡混匀后直接进行核酸提取。
1.3 核酸提取 取1.2处理的标本200μl,采用德国Qiagen公司RNeasy Mini Kit试剂(批号74104)在QIA Cube核酸提取仪上对标本进行RNA的提取,最终RNA洗脱体积为50μl。
1.4 荧光定量 RT-PCR(Real-Time RT-PCR)检测 首先采用通用型EV核酸荧光定量RT-PCR试剂盒对待检核酸进行检测,然后对阳性标本进行EV-A71型和 CV-A16型核酸检测,最后对非 EVA71非CV-A16的EV核酸阳性标本,使用CV-A6型荧光定量RT-PCR试剂盒进行核酸检测,鉴定出CV-A6型阳性标本。该检测均使用江苏硕世生物科技有限公司试剂盒,在ABI 7500荧光定量PCR仪上进行,反应体系、扩增条件和结果判读均按照试剂盒说明书进行。
1.5 CV-A6 VP1区扩增和测序 采用传统RTPCR方法,将1.4鉴定为CV-A6型阳性的核酸标本进行扩增,选用德国Qiagen公司的One step RT-PCR Kit试剂(批号210212),在ABI公司的veriti PCR仪上进行扩增,反应体系、扩增参数和引物序列参见文献[2],扩增阳性产物纯化后送上海伯杰生物科技有限公司进行双向测序。
1.6 序列拼接和进化分析 应用Lasergene7.1软件对序列进行拼接,从GenBank下载相关CV-A6型VP1区的核苷酸序列,利用MEGA6.0软件与本研究完成测序的序列进行比对,应用Neighbor-Joining(N-J)法构建进化树。
2 结果
2.1 基本情况 沈阳地区2013—2017年共采集手足口病患者临床标本2 881份,肠道病毒核酸阳性标本2 066 份,阳性率为71.71%(2 066/2 881)(表1)。 其中 EV-A71 占 33.30%(688/2 066),CV-A16占24.20%(500/2 066),其他肠道病毒占 42.50%(878/2 066)。其中2015年其他肠道病毒比例大幅度增加,达到83.19%(292/351)。
2.2 CV-A6检测情况 本实验室对878份非EVA71非CV-A16的其他肠道病毒阳性标本进行CVA6核酸检测,共检测到CV-A6阳性标本575份,占总肠道病毒阳性标本的构成比为27.83%,在手足口病病原谱中仅次于EV-A71(33.30%),超过CVA16居第2位。表1可见,2013—2017年中CV-A6所占总肠道病毒的构成比间差异具有统计学意义,其中 2015年所占比例最高达到 68.38%(240/351),CV-A6成为该年的绝对优势流行株;而高峰后的2016年则为5.65%,降低到近年最低。
2.3 RT-PCR扩增和序列测定 对荧光定量RTPCR检出的CV-A6核酸阳性标本575份,本研究按照不同年份每年随机抽取16份,连续5年间共计80份标本,使用传统RT-PCR方法扩增CV-A6的全长VP1区,除4份标本因病毒载量较低导致PCR产物浓度不足以进行测序反应外,其余76份样本成功扩增,PCR产物与预计片段大小相符。
2.4 CV-A6系统进化树分析 为分析沈阳地区CV-A6型分子流行病学特征,从GenBank数据库中选取国内各省(市)和其他国家或地区具有典型代表性的CV-A6全长VP1区序列共65条,分别来自法国、西班牙、美国、日本、芬兰,以及中国(山东、吉林、辽宁、四川、河南、江西、河北、湖南、云南、广东、福建和台湾),同本研究测得的76条序列进行遗传进化分析,并构建系统进化树(图1)。从进化树图可以看出,141条CV-A6 VP1区序列在系统进化树中分为4个不同基因型(A、B、C和D),各基因型间核苷酸差异在14%~23%之间,各基因型间具有明显的时空分布差异。A基因型只包括1949年分离自美国的原型株Gdula株(AF081297),B基因型仅包括4株1992—2007年在中国山东和广东发现的病毒株(192022、KP143075、AFP051 和 KP142078),而C基因型也仅有2株分别为1996年中国山东株(JQ364887)和 2008 年的印度株(JN203517)。1999—2017年在中国、日本、芬兰、法国和西班牙发现的134株病毒株则一起构成了D基因型的进化分支。
D基因型又可以进一步分为3个基因亚型(D1、D2和D3)。 D1亚型于1999—2008年间分别在日本(1999年)、西班牙(2008年)和法国(2010年)发现;D2亚型在2006—2013年间流行于中国和日本;D3亚型毒株则是于2009—2017年间分别由中国、日本、芬兰、法国和西班牙陆续发现。进化分析表明,D3亚型由两个分支D3a和D3b构成,其核苷酸差异在4% ~8%,其中D3b进化分支在2008—2013年分别流行于西班牙(2008年)、芬兰(2008年)、法国(2010 年)、日本(2011 年)和中国(2011—2013年),其余GenBank下载的毒株和本研究分离株序列共计101条均属于D3a进化分支;进化分析表明,随时间推移该分支又进化成两个小分支分别为D3a.1和D3a.2,D3a.1流行空间广持续时间短,主要于2010—2013年间流行于法国(2010年)和中国(2010—2013年),同源性在97% ~99%;D3a.2流行空间局限但持续时间长,除2009—2010年间中国台湾的2株外,其余均为2012—2017年间的中国流行株,同源性在95% ~99%之间。
3 讨论
近年监测结果显示,我国手足口病的流行高峰、流行强度和病原构成等主要流行特征在高、中、低纬度地区间差异较大[1]。东北地区处于我国高纬度,与处于中纬度的安徽、上海等地相比流行高峰出现晚、流行强度低、呈明显单峰分布,病原更新速率也较慢,本地区的手足口病具有独特的流行特点与病原进化特征。
2008年,芬兰首次报道CV-A6导致手足口病的暴发,在欧洲、亚洲、美洲和大洋洲相继出现了不同程度的流行[3-7],在世界范围内逐渐成为手足口病的主要病原。中国与其他国家相比CV-A6导致手足口病的报道较晚,有关监测表明2011年的广东和福建等低纬度地区、2012年的江苏和上海等中纬度地区、2013年的西北和华北等高纬度地区相继有CV-A6导致手足口病的报道[8-12],总体呈现自2011年开始由南向北迅速流行的趋势。沈阳地区最早是在2013年手足口病监测中发现CV-A6毒株,历年监测结果表明该型毒株占肠道病毒的总构成比达27.83%,在手足口病的病原谱中仅次于EV-A71,超过CV-A16居第二位。2013—2017年的连续监测中,CV-A6型在所有手足口病肠道病毒各型别的构成比变化较大,2015年是近几年监测中手足口病肠道病毒阳性率最高的年份,其阳性率接近80%,病原构成中CV-A6型达到68.38%成为该年的绝对优势流行株。尤其值得关注的是,该年有16例手足口病重症患者经本实验室确诊是由CV-A6型毒株引起的,在以往监测中鲜有所见,说明该型毒株在进化过程中传播力增强的同时,致病力也有所改变。

◆沈阳序列图1 CV-A6型肠道病毒VP1基因核苷酸全长序列系统进化树◆Shenyang sequencesFig.1 Phylogenetic tree based on the entire VP1 nucleotide sequences of the CV-A6
VP1蛋白是肠道病毒最重要的衣壳蛋白,含有抗原决定位点和受体识别表位,具有与病毒血清型对应的遗传多态性,因此VP1区是肠道病毒基因分型和遗传进化分析的重要研究对象[13]。本研究参照文献[2]根据VP1构建CV-A6型的系统进化树,将其划分为A、B、C、D等四个不同基因型,从图1可以看出各基因型具有显著的时空分布与进化规律。2007年之前,国内发现的5例CV-A6型病毒株均为非D型,并且仅局限在广东和山东等少数省份(山东2株分别为A型和C型、广东3株均为B型),两省都为沿海发达省份,与境外往来较多,推测其均为输入型病例,但由于非D型毒株的自身遗传特点和宿主免疫屏障等原因,其在两地未能进行有效的传播。从2008年开始,国内发现的CV-A6型毒株均为D型,根据进化树可以看出其在我国的进化大致可以分为三个阶段,第1个阶段发生在2009年之前以D2基因亚型流行为主;第2阶段2009—2012年为进化的过渡阶段,D2和D3亚型共存,并主要以D3a分支为主,D3b毒株仅在小范围内流行;第3阶段为2013年以后,以D3a分支为主的CV-A6型毒株在国内全面爆发流行。以上可见,我国发现CVA6型毒株以来主要以D2和D3亚型流行为主,且以D3a分支多见。
进一步研究发现,D3a分支的CV-A6型毒株进化形成2个独立分支,本研究将其命名为D3a.1和D3a.2,2分支间核苷酸差异在4% ~8%间。从图1可以看出,2013年以来沈阳地区的CV-A6型全部位于D3a上,且以D3a.2为主。从时间来看,D3a.1分支仅包括2013年和2014年毒株,而2015—2017年间的所有毒株均位于D3a.2分支上(部分2013年和2014年毒株也位于该分支上)。可见,从时空来看,CV-A6型的D3a.2分支毒株已于2015年传入沈阳地区,但此时沈阳地区人群针对D3a.2分支毒株的基础免疫保护水平极低导致普遍易感,这也是沈阳地区在2015年手足口病检测阳性率近年最高(79.59%),且CV-A6型居肠道病毒所有型别首位(68.38%)的原因。
本研究通过针对沈阳地区2013—2017年手足口病中CV-A6的检测和基因进化特征分析,进一步客观的证实了CV-A6是手足口病的重要病原体之一,阐明了2015年CV-A6在沈阳暴发流行的病原学基础。手足口的监测和防控应该采取多病原和多地区联动的措施,加强对新出现或新传入的毒株类型的监测,提高警惕、避免造成严重的疾病负担和公共卫生事件。
利益冲突 无
