一种新颖的识别酶EC编号的计算方法
2018-10-15崔浩
崔浩
(上海海事大学信息工程学院,上海 201306)
0 引言
酶是一种可以加速化学反应的分子。大多数的酶是蛋白质并且参与几乎所有的代谢过程,以创造出足够的能量来维持生命。为了标记酶,每个酶会分配一个酶委员会编号(Enzyme Commission(EC)number),简称EC编号。EC编号由四个数字组成,例如1.1.1.1。详细地说,前三个数字代表酶可以参与的化学反应类型,最后一个数字表示底物专一性或编号[1]。到目前为止,EC编号已被用在多个公共数据库。例如,在京都基因与基因组百科全书(KEGG)[2]中,在代谢途径中的化学反应被至少一个EC编号打上标签,用来表示哪种酶可以催化这种反应。
识别一个所给酶的EC编号对揭示其功能是相当重要的,研究人员可以进一步推断出这个酶可以参与哪种类型的反应。然而,通过传统的实验来获取所给酶的EC编号是耗时和昂贵的。建立计算方法来推断酶的EC编号是一种可替代的方法,这样可以充分利用几个已知的信息来给出有用的提示。到目前为止,在这方面有人做出了一些努力。然而,他们中的大多数都集中在预测酶所属EC编号的前两个数字,甚至是第一个数字。在这方面的首次研究是由Jensen等人提出的[3],他们使用各种序列相关物理化学特征来表示酶以及人工神经元网络作为预测引擎。此后,许多在这方面的预测方法相继被提出。在这些方法中,酶总是由几种类型的特征来表示,如氨基酸组成[4]、蛋白质功能域组成[5]、伪氨基酸组成[6,7]、蛋白质结构[8]、基因本体[9],以及采用经典的机器学习算法,如人工神经元网络[3]、支持向量机[10]、贝叶斯[11]、最近邻算法[12],来建立预测模型。虽然这些方法能够产生良好的性能,但是它们不能准确地判定酶的整个EC编号。此外,以前的方法只考虑了酶的信息,从而引起了方法的局限性。
在本文中,我们构造了一种新的分类器来识别酶的EC编号。为了训练这个分类器,所有的酶以及它们的EC编号都是从ENZYME数据库中检索出来的[13]。不同于以前把酶分为几类的方法,在本文中是把一个酶和它的所属EC编号配对为一个正样本。负样本随机被产生并且产生的个数和正样本的个数一样多。然后,酶的EC编号的确定问题被转换成一个二分类问题,即,测定一个酶和一个EC编号是否可以配对。从蛋白质相互作用数据库STRING(https://string-db.org/,version 10.0)里获得的蛋白质-蛋白质相互作用被用来测量任何两个酶之间的相似性[14],并提出了一种新的方案来评估两个EC编号之间的关系。通过集成上述两种类型的关系,可以评估两个样本之间的关系,样本之间的关系被采用作为基于支持向量机的分类器里的核函数。在五个不同的数据集上应用提出的基于5-折交叉验证的分类器,得到的整体准确率为0.810,马修斯相关性系数为0.629,F1-measure为0.791。相信所提出的方法是一个识别酶的EC编号的有用工具。
1 材料和方法
1.1 材料
酶和酶的EC编号是从ENZYME数据库的站点(http://enzyme.expasy.org/,2016年2月访问)检索得来的[13]。在这里,我们只考虑了人类的酶和它们对应的EC编号。为了构建一个二分类器,把一个EC编号C分配给酶E,那么它们被配对为一个样本,记为S=(E,C)。因为我们使用了蛋白质-蛋白质相互作用来评估酶之间的相似性,所以没有蛋白质-蛋白质相互作用信息的酶对会被丢弃,从而产生了1,480对酶和EC编号。这些对在本研究中被称为正样本。
为了评估二分类器的性能,负样本是必要的。在这里,我们随机配对酶和EC编号作为负样本。但是,它们不能是正样本。尽管一些负样本可能是实际的酶和EC编号对,但我们仍然将它们用作负样本,因为这种类型的样本非常少,并且它们不会对预测结果产生很大影响。为了充分评估所提出的分类器,我们随机产生了5组负样本,每组包含与正样本个数一样多的样本。每组负样本和正样本都组成一个数据集,其他组负样本也一样和正样本组成数据集,即我们构建了五个数据集来评估分类器,分别记为D1,D2,D3,D4,D5。
1.2 蛋白质-蛋白质相互作用
蛋白质-蛋白质相互作用是研究蛋白质相关问题的有用信息[15,16]。几项研究表明,可以相互作用的蛋白质更可能共有共同的功能。如第1节所述,大多数酶都是蛋白质。使用蛋白质-蛋白质相互作用来评估酶之间的联系是可行的。
在本文中,我们使用了在STRING(https://stringdb.org/,版本10.0)中报告的蛋白质-蛋白质相互作用,这是一个集成了被验证和预测的蛋白质-蛋白质相互作用的公共数据库,这些相互作用源自(I)基因组上下文预测;(II)高通量实验;(III)保守性共表达;(四)自动文本挖掘;(五)数据库先验知识。因此,他们可以广泛地评估蛋白质之间的关系,并已应用于研究许多生物问题[15,17]。我们从文件“9606.protein.links.v10.txt.gz”中提取了人类蛋白质-蛋白质相互作用。每个相互作用包含两个蛋白质和一个分值,蛋白质以Ensembl IDs表示,分值表示相互作用的强度。为了公式化表达,我们记蛋白质 p1和 p2之间的一个相互作用分值为S(p1,p2)。因为相互作用分值的范围在150到999之间,所以我们评估了酶E1和E2之间的相似性为:

1.3 EC编号之间的相似性
如第1节所述,以前的大多数方法只考虑了构建分类器的酶的信息。在这里,我们给出了一个新的方案来评估两个EC编号之间的关系,这将进一步用于构建分类器。
对于任何EC编号C=W.X.Y.Z,它被转换为由四个元素组成的集合,公式化为S(C)={W,W.X,W.X.Y,W.X.Y.Z}。然后,给定两个EC编号,比如说C1和C2,它们的相似性可以被转化为两个集合S(C1)和S(C2)之间的关系,由公式表达为:

1.4 基于支持向量机的分类器
在1.2节和1.3节中,评估了酶之间的相似性(参见方程1)和EC编号之间的相似性(参见方程(2))。显然,通过集成它们,可以评估任意两个样本(酶和EC编号对)S1=(E1,C1)和S2=(E2,C2)之间的相似性,相似性被定义为:

很容易看出,Q(S1,S2)值的范围在0到1之间。Q(S1,S2)的值越高,意味着S1和S2的相似性越高。
通过使用方程(3)作为核函数,可以构建基于支持向量机的分类器来识别酶的EC编号。假设Dt是一个包含m个样本的训练集,比如说S1,S2,…,Sm,对于每个样本Si(1 ≤i≤m ),它可以通过其与Dt里所有样本的相似性来表示,其他的样本也是如此,即:
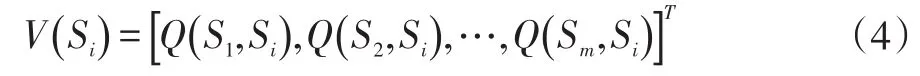
然后,采用经典的支持向量机算法并对数据集Dt进行训练,从而生成分类器F.对于任何测试样本S,可以表示为:

测试样本的类别要么为正样本要么为负样本,能够由分类器F来预测。
本文使用了著名的开源机器学习以及数据挖掘软件Weka,Weka软件收集了一套用于数据挖掘任务的机器学习算法。其中一个名为“SMO”的工具实现了一种支持向量机。“SMO”工具使用了John Platt的连续最小优化算法优化支持向量机的训练过程。为了快速实现基于支持向量机的分类器,本文采用了“SMO”工具,并使用其默认参数执行。
1.5 分类器评价
本文研究的是二元分类问题,所以由分类器产生的预测结果可以统计为一个2×2的混淆矩阵M,公式化表达为:

其中矩阵包含4个值:TP代表将正样本预测为正样本的数量,FN代表将正样本预测为负样本的数量,FP代表将负样本预测为正样本的数量,TN代表将负样本预测为负样本的数量。
基于混淆矩阵M中的四个值,我们还计算出其他对分类模型的评价指标。以下总共计算了7个指标,分别为灵敏度(SN)、特异度(SP)、准确率(ACC)、马修斯相关性系数(MCC)、精确率(Precision)、召回率(Re⁃call)、F1-measure,计算公式分别为:

所有上述指标均用于评估本研究中提及的任何分类器的性能,其中准确率,马修斯相关性系数和F1-measure是更重要的指标,因为它们可以测量分类器整体的性能,其他评价指标(灵敏度、特异度、精确率、召回率)也会给出,以供读者参考。从公式中不难看出,灵敏度和召回率的公式是一样的,所以在文章中评价分类器性能的时候,我们只需展示灵敏度的结果就可以了。
2 实验结果与讨论
2.1 基于支持向量机的分类器与其他三种分类器的结果比较
在本文中,我们提出了基于支持向量机的分类器识别酶的EC编号。整个分类器的构造和评估的流程如图1所示:
为了表明基于支持向量机分类器的有效性,本文还使用了其他三种经典的机器学习算法:贝叶斯网络,JRip和随机森林来构建分类器,通过比较结果来说明基于支持向量机分类器的有效性。为了方便起见,我们采用了 WEKA 中的“SMO”、“BayesNet”、“Jrip”和“RandomForest”工具分别实现这四个分类器,工具都使用其默认参数执行,测试流程都按照图1所示进行,只有分类器不同而已。
本小节给出了所提出的四种分类器的测试结果。如第2.1节所述,构建了五个数据集D1,D2,D3,D4,D5。对于每一个数据集,执行基于支持向量机的分类器,并通过5-折交叉验证来评估其性能。预测结果由灵敏度、特异度、准确率、马修斯相关性系数,精确率和F1-measure来表示,如表1所示:

图1 分类器的构造和评估

表1 基于支持向量机的分类器在5个数据集上的性能
对于第2.1节中提到的五个数据集,它们都用于测试基于贝叶斯网络,JRip和随机森林的分类器的性能,通过5-折交叉验证来进行评估。预测结果还是由灵敏度、特异度、准确率、马修斯相关性系数,精确率和F1-measure来表示,如表2~4所示:

表2 基于贝叶斯网络的分类器在5个数据集上的性能

表3 基于JRip的分类器在5个数据集上的性能
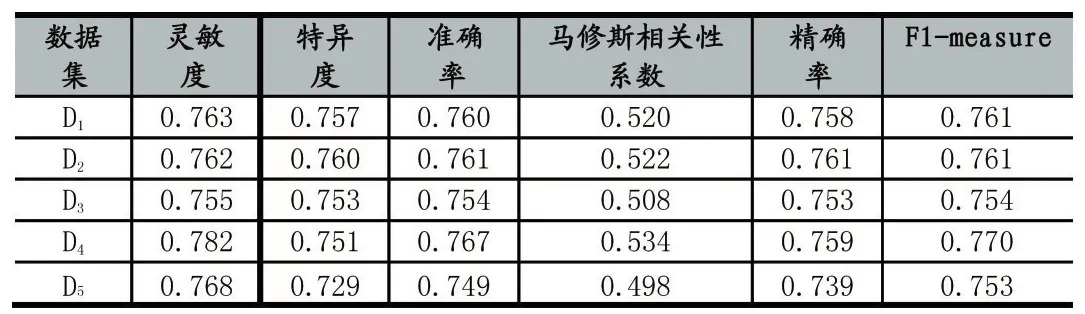
表4 基于随机森林的分类器在5个数据集上的性能
表1~4不容易直观地看出每个分类器每个评价指标的值的分布规律,也不能直观地做出分类器之间的比较,所以我们根据表中的数据画出了四张箱形图,如图2所示。
从图2中我们可以看出,基于支持向量机的分类器提供了最好的指标。对于灵敏度而言,基于随机森林的分类器产生了最高的值,但是它却提供了最低的特异度;对于特异度而言,基于贝叶斯网络的分类器产生了最高的值,但是它却提供了最低的灵敏度;证明它们不如基于支持向量机的分类器。这也可以通过观察这三个分类器的准确率,马修斯相关性系数和F1-mea⁃sure来证明。
为了进一步比较这三个分类器和基于支持向量机分类器的性能,在表中列出了每个分类器产生的每个指标的平均值,$标出每列平均值的最大值,如表5所示:

图2 基于四种算法的分类器的评估指标结果的箱形图
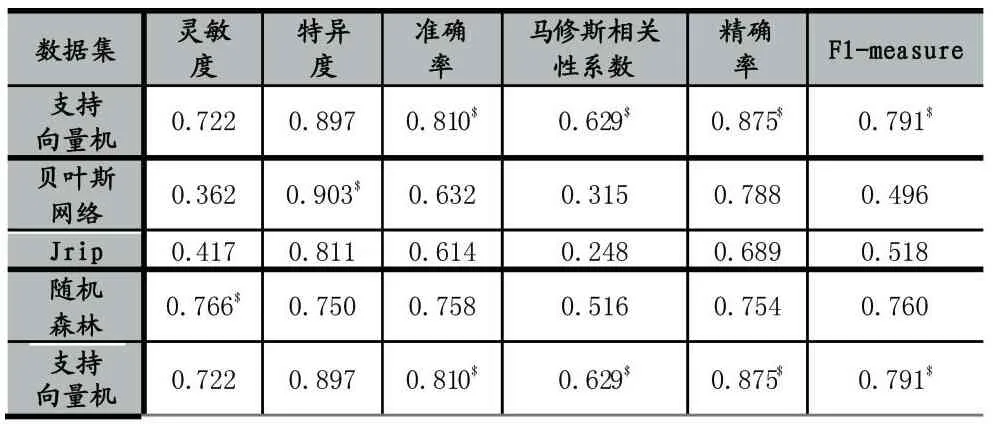
表5 基于四种算法的分类器的性能的评估指标平均值结果比较
可以看出,平均值的比较结果和图2的比较结果趋势是一样的,同图2解释,这里就不再赘述。稍微值得一提的是基于支持向量机的分类器的灵敏度,特异度和精确率分别为0.722,0.897和0.875,表明了该分类器为正样本和负样本的预测提供了很高的准确率,并且在预测为正的样本中,大多数是正确的。对于能够评估该分类器整体性能的准确率,马修斯相关性系数和 F1-measure,它们的值分别是 0.810,0.629和0.791,这表明该分类器在识别酶的EC编号方面有着良好的性能。为了进一步表明提出的基于支持向量机的分类器比其他三个分类器优越得多,由基于支持向量机的分类器产生的准确率(马修斯相关性系数和F1-measure)比由其他分类器获得的准确率(马修斯相关性系数和 F1-measure)至少高出 0.05(0.11,0.03)。
此外,每个分类器产生的每个指标的标准差也列了一个表格,标准差是由表1~4中的数据计算所得,$标出每列标准差的最小值,如表6所示:
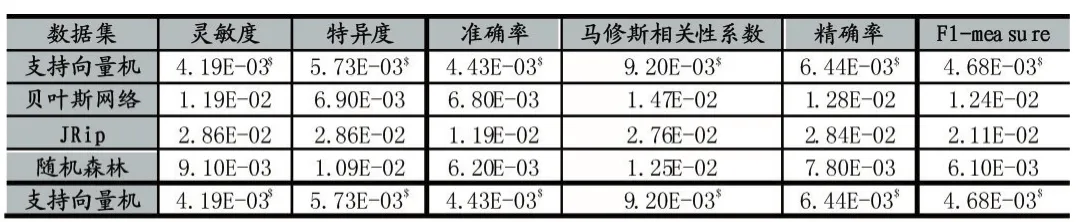
表6 基于四种算法的分类器的性能的评估指标标准差结果比较
从表中可以观察到,基于支持向量机分类器的所有指标的标准差都是最小的且都小于0.01,这意味着尽管负样本在五个数据集中不同,但是该分类器的性能是相当稳定的。所有的这些证据都意味着基于支持向量机的分类器比其他三个分类器都要强大。
2.2 调整参数α
本文除了通过比较基于四种算法的分类器说明支持向量机的算法更具优势外,还在此基础上改善基于支持向量分类器的预测准确度,那就是调整2.3节中计算EC编号之间相似性的公式2中的参数α,我们尝试过{0,0.1,0.2,0.3,0.4,0.5,0.6,0.7,0.8,0.9,1,1.5,2,2.5},分类器性能评价6个指标具体数据就不在这里用表格详细展示了,我们挑出了具有代表性的评价指标马修斯相关性系数MCC来展示各参数在2.1节中提到的5个数据集上的结果,如图3所示:
从图中的MCC平均值可以看出参数α越小,MCC平均值越大,说明当参数α为0时,预测的准确率相比之下最高。5个不同数据集在参数相同的情况下MCC值差异很小,说明即使负样本不同,也不会影响到分类器的性能。还有一个很明显的现象,那就是参数α从0到1时MCC值是缓慢递减的,而从α大于1时,MCC值相比之下递减速度更快。

图3 马修斯相关性系数在不同参数下的结果
2.3 分类器的适用性
在本文中,我们建立了一个基于支持向量机的分类器来识别酶的EC编号。然而,识别酶的EC编号是否特殊?这小节将证明所提出的分类器是针对这个问题的,这表明在2.1节中提到的结果是相当可靠的。
我们随机地生成了1,480个酶和EC编号对作为正样本和1,480个酶和EC编号对作为负样本,这些对组成了一个数据集。同理,随机生成其他四个数据集。因此,生成了五个数据集,记为然后,对这些数据集分别执行基于支持向量机,贝叶斯网络,JRip和随机森林的分类器,通过5-折交叉验证进行评估。预测结果也被统计为2.1节中提到的6个指标,为了方便观察四种分类器在每个评价指标下的每个数据集上的结果比较,我们画出了6张对应的图,如图4所示。
可以观察到,尽管不同分类器在相同数据集上获得的灵敏度和特异度差异很大,但是其他四个指标几乎是在同一级别,特别是对于准确率、马修斯相关性系数和F1-measure。所有这些都表明了基于支持向量机的分类器在这种情况下并不优于其他三种分类器,这意味着基于支持向量机的分类器可以捕获D1,D2,D3,D4,D5里正、负样本中的关键差异。此外,通过观察由基于支持向量机的分类器所产生的准确率和马修斯相关性系数,它们的值分别都在0.5和0左右,这表明预测结果与通过随机预测获得的结果非常相似。这是合理的,因为所有的样本都是随机产生的,这意味着它们之间的差异很小。

图4 四种分类器在每个评价指标下的每个数据集上的结果比较
3 结语
本文提出了基于支持向量机的二分类器来识别酶的EC编号,与以往仅考虑酶的信息和将酶分成若干类的问题的研究不同,本文将酶和EC编号配对作为样本,把问题转化成了二分类问题。为了构建分类器,酶之间的关系与EC编号之间的关系被集成。测试结果表明了基于支持向量机的分类器对于识别酶的EC编号是非常有效的。希望该分类器可以成为一种将EC编号分配给新型酶的新工具,并且分类器的构建思想可以提供新的见解,从而为涉及多层分类的问题建立更好的预测模型。
