意大利蜜蜂工蜂中肠发育过程中长链非编码RNA的差异表达分析
2018-10-11郭睿耿四海熊翠玲郑燕珍付中民王海朋杜宇童新宇赵红霞陈大福
郭睿,耿四海,熊翠玲,郑燕珍,付中民,王海朋,杜宇,童新宇,赵红霞,陈大福
意大利蜜蜂工蜂中肠发育过程中长链非编码RNA的差异表达分析
郭睿1,耿四海1,熊翠玲1,郑燕珍1,付中民1,王海朋1,杜宇1,童新宇1,赵红霞2,陈大福1
(1福建农林大学蜂学学院,福州 350002;2广东省生物资源应用研究所,广州 510260)
【目的】长链非编码RNA(lncRNA)在真核生物的基因表达、表观遗传和细胞周期调控等方面发挥重要功能。本研究旨在探究意大利蜜蜂(, 简称意蜂)工蜂中肠发育过程中lncRNA的表达谱及其作用。【方法】利用RNA-seq技术和链特异性建库方法对意蜂7和10日龄工蜂中肠(Am7、Am10)进行深度测序,下机的原始数据经过Perl脚本过滤得到高质量有效读段。利用bowtie工具将有效读段比对核糖体数据库,进一步利用TopHat2软件将未比对到核糖体数据库上的数据比对到参考基因组。利用CPC和CNCI软件对转录本的编码能力进行预测。通过RT-PCR对部分lncRNA进行鉴定。利用edgeR软件进行差异表达lncRNA(DElncRNA)分析,进而预测lncRNA的上下游基因,并对上下游基因进行GO及KEGG代谢通路富集分析。联用RNAhybrid、Miranda和TargetScan软件预测DElncRNA靶向结合的miRNA及miRNA靶向结合的靶基因,并通过Cytoscape软件构建DElncRNAs-miRNAs-mRNAs的调控网络。最后,通过RT-qPCR验证测序数据的可靠性。【结果】Am7和Am10的深度测序分别获得134 802 058和147 051 470条原始读段,经严格过滤分别得到134 166 157和146 293 288条有效读段;共得到3 890个DElncRNA,包括2 005个上调lncRNA与1 885个下调lncRNA。RT-PCR验证结果显示共有8个lncRNA能扩增出符合预期的目的片段,表明预测出的lncRNA真实存在。DElncRNA的上下游基因数为1 793个,它们涉及42个GO条目,包括代谢进程、发育进程、细胞进程、应激反应和免疫系统进程等;这些上下游基因还涉及251条代谢通路,包括碳代谢、嘌呤代谢和脂肪酸的生物合成等物质代谢通路,硫代谢、甲烷代谢和氧化磷酸化等能量代谢通路,Hippo信号通路、Wnt信号通路和Notch信号通路等信号通路,溶酶体、内吞作用和泛素介导的蛋白水解等细胞免疫通路,以及MAPK信号通路、Jak-STAT信号通路和NF-kappa B信号通路等体液免疫通路,上述结果表明DElncRNA在意蜂中肠发育过程中参与物质和能量代谢、细胞生命活动和免疫调控。进一步分析发现TCONS_00020918可通过调控西方蜜蜂王浆主蛋白1编码基因在意蜂工蜂中肠的营养吸收、级型分化中发挥功能。DElncRNA的调控网络分析结果显示DElncRNA与miRNA、mRNA间存在复杂的调控关系,部分DElncRNA处于调控网络的中心位置且能靶向结合较多的miRNA,也有部分miRNA可被多个DElncRNA共同靶向,表明这些DElncRNA可能在中肠发育中发挥重要作用。随机挑取5个DElncRNA进行RT-qPCR验证,结果显示它们的表达量变化趋势与测序结果一致,证实了本研究测序数据的可靠性。【结论】差异表达长链非编码RNA(DElncRNA)广泛参与意蜂工蜂中肠的新陈代谢、细胞活动和免疫调控并作为竞争性内源RNA(ceRNA)发挥作用,研究结果为关键lncRNA的筛选和功能研究提供了必要的数据支持。
意大利蜜蜂;中肠;发育;长链非编码RNA;上下游基因
0 引言
【研究意义】蜜蜂作为社会学模式昆虫,在发育学、行为学和神经生物学等方面具有重要价值[1]。同时,蜜蜂也是最重要的授粉昆虫,具有不可替代的经济价值和生态价值[2]。意大利蜜蜂(,简称意蜂)具有优越的采集能力、造脾能力和分泌蜂王浆能力[3],在世界各地的养蜂生产中广泛使用。目前,有关蜜蜂中肠发育机理及调控机制的研究极为滞后,非编码RNA(non-coding RNA,ncRNA)在中肠发育过程中的作用的相关信息也十分有限。在转录组水平对长链非编码RNA(long non-coding RNA,lncRNA)在意蜂工蜂中肠发育中的作用进行研究,可为深入解析意蜂工蜂中肠发育的分子机理提供新的思路和线索。【前人研究进展】随着测序技术的发展,起初被认为基因转录“噪音”的ncRNA成为近几年的研究热点。在人类基因组中,编码蛋白序列所占比例不到2%,超过98%的序列是不编码蛋白质的[4]。ncRNA根据其长短和形状分为小RNA、lncRNA和环状RNA等。其中,lncRNA已被证明在剂量补偿效应[5]、表观遗传[6]、细胞周期[7]和细胞分化调控[8]等众多生命活动中发挥重要作用,因此成为生命科学领域的研究热点。lncRNA的作用方式多样,包括作为信号分子调控上下游基因转录[9],作为诱导分子充当分子阻断剂[10],作为引导分子与蛋白结合[11],作为支架分子发挥“脚手架”作用[12],作为小RNA的前体[13],以及作为竞争内源RNA结合miRNA对mRNA起剪接调控作用[14]。近年来,有关lncRNA在肿瘤发生机制方面作用的研究取得了显著进展,在脂肪代谢、肌肉发育及免疫抗病等机理方面作用的研究也取得了许多突破性进展[15-16]。然而,lncRNA在昆虫领域的相关研究较为滞后,仅在果蝇和家蚕等极少数模式昆虫中有较少的lncRNA相关研究报道[17]。此前,有研究报道lncRNA在蜜蜂的级型分化[18]、卵巢发育[19-20]和抵抗病毒入侵[21]中起重要的调控作用,但总体上有关蜜蜂lncRNA的研究仍非常有限。肠道是昆虫的重要消化器官和免疫器官。蜜蜂肠道的相关研究主要集中在肠道微生物方面[22-27],如Engel等[26]利用二代测序技术对意蜂工蜂后肠进行宏基因组测序,基于数据分析发现意蜂后肠内的少量细菌种类存在极大的遗传多样性,进一步的比较分析发现不同的细菌种类涉及不同的功能,包括宿主互作、菌膜形成及碳水化合物水解等。【本研究切入点】蜜蜂中肠发育机理及调控机制仍不明确,关于lncRNA在蜜蜂中肠发育过程中作用的研究未见报道。【拟解决的关键问题】结合RNA-seq技术和链特异性cDNA建库方法对意蜂7和10日龄的工蜂中肠进行测序,对中肠发育过程的lncRNA进行差异表达分析,进一步通过上下游基因分析和lncRNA-miRNA-mRNA调控网络分析DElncRNA在意蜂工蜂中肠发育过程中的作用。
1 材料与方法
试验于2017年在福建农林大学蜂学学院蜜蜂保护实验室进行。
1.1 生物材料
供试意大利蜜蜂工蜂取自福建农林大学蜂学学院教学蜂场。
1.2 样品制备与Illumina测序
从群势较强的意蜂蜂群中选取有封盖子的巢脾,快速提至实验室,放入(34±0.5)℃培养箱中培养至工蜂出房,将刚出房的工蜂(记为0 d)放入干净的四周打孔以通风的塑料盒中,盒子上方插入一支装有50%(w/v)无菌糖水的饲喂器(图1),(34±0.5)℃条件下恒温培养,每日检查工蜂存活情况,及时清理死亡的工蜂。进行3次生物学重复。

图1 意蜂工蜂的人工饲养
选取意蜂7和10日龄工蜂中肠作为测序材料。分别快速拉取意蜂7日龄工蜂中肠(Am7)和10日龄工蜂中肠(Am10),液氮速冻后置于-80℃超低温冰箱保存备用,每只中肠的取样时间严格控制在15 s以内。首先用RNA抽提试剂盒(AxyPrepTMMultisource Total RNA Miniprep Kit)(TaKaRa公司,日本)抽提蜜蜂中肠样品的总RNA,为最大限度地保留所有非编码RNA(ncRNA),去除核糖体RNA后的mRNA和ncRNA用裂解缓冲液随机打断为小片段,作为模板用六碱基随机引物、缓冲液、dNTPs、RNase H和DNA polymerase I合成cDNA第二链。经过QiaQuick PCR试剂盒(Qiagen公司,德国)纯化并加EB缓冲液洗脱经末端修复、加碱基A,加测序接头,然后通过尿嘧啶-N-糖基化酶(UNG)降解cDNA第二链。消化产物经琼脂糖凝胶电泳和PCR扩增,PCR产物经高碘酸钠(Sigma 公司,美国)处理。委托广州基迪奥生物科技有限公司对上述cDNA文库进行双端(paired-end)测序,测序平台为Illumina HiSeq 4000。测序数据已上传NCBI SRA数据库,BioProject号:PRJNA406998。
1.3 高通量数据分析
1.3.1 LncRNA的生物信息学预测 对于下机数据,利用Perl脚本去除含有adaptor、未知核苷酸比例>10%和低质量reads,获得有效读段(clean reads)。使用短reads比对工具bowtie[28]分别将样品Am7-1、Am7-2、Am7-3、Am10-1、Am10-2和Am10-3的clean reads比对(mapping)到核糖体数据库(最多允许5个错配),去除mapping上核糖体的reads,利用TopHat2软件[29]将保留下来的数据进一步比对西方蜜蜂的参考基因组(Amel_4.5)[30]。利用FPKM(Fragments Per Kilobase of transcript per Million mapped reads)法计算基因表达量。利用R软件(version 2.16.2)计算各样品之间的相关性系数。
lncRNA与mRNA相比在序列保守型、ORF长度等特点上具有一定差异,利用软件CPC[31]和CNCI[32]对新转录本的编码能力进行预测,选取二者的交集作为可靠的预测结果。
1.3.2 DElncRNA及其上下游基因分析 利用edgeR软件[33]进行lncRNA的差异分析,得到DElncRNA。LncRNA的功能与其编码基因位置临近的蛋白编码基因关系密切,位于上游的lncRNA可能与启动子或共表达基因的其他顺式作用元件存在交集,从而在转录或转录后水平进行基因表达调控;位于3′ UTR或基因下游的lncRNA可能参与其他调控作用。因此,对lncRNA进行注释,如果其位于一个基因的上游或下游,这些lncRNA有可能与顺式作用元件所在区域有交集,从而参与转录调控。利用Omicshare平台(http://www.omicshare.com/tools/index.php/)对DElncRNA的上下游基因进行GO(Gene Ontology)分类和KEGG pathway富集分析。
1.3.3 DElncRNA的调控网络构建 LncRNA可以竞争结合miRNA,从而减少miRNA结合mRNA,表明lncRNA可以通过miRNA调控mRNA的表达。利用RNAhybrid[34]、Miranda[35]和TargetScan[36]软件预测DElncRNA靶向结合的miRNA,以及miRNA结合的mRNA,根据上述靶向结合关系构建lncRNA-miRNA- mRNA的调控网络。利用Cytoscape软件[37]对上述调控网络进行可视化。
1.4 lncRNA的RT-PCR验证
随机选取9个lncRNA,根据它们的序列利用DNAMAN软件设计相应的特异性引物。委托上海生工生物工程有限公司合成引物。利用RNA抽提试剂盒(TaKaRa,中国)提取Am7和Am10的总RNA,作为模板进行反转录,得到的cDNA作为模板进行PCR。PCR反应体系(20 μL)包括cDNA模板1 μL,上游引物1 μL,下游引物1 μL,Mixture 10 μL,无菌水补至20 μL。PCR程序:94℃预变性5 min;94℃变性50 s,55℃退火30 s,72℃延伸50 s,共34个循环;72℃再延伸10 min。PCR产物经1.5%琼脂糖凝胶电泳和凝胶成像仪(上海培清,中国)检测。
1.5 DElncRNA的实时荧光定量PCR(RT-qPCR)验证
为了验证RNA-seq数据的可靠性,随机选取5个DElncRNA进行RT-qPCR验证。利用DNAMAN软件设计相应的特异性引物。委托上海生工生物工程有限公司合成引物。利用RNA抽提试剂盒(TaKaRa,中国)提取Am7和Am10的总RNA,作为模板进行反转录,得到的cDNA作为模板进行PCR。RT-qPCR反应按照SYBR Green Dye试剂盒(Vazyme公司,中国)操作说明书进行,每个反应进行3次重复。反应体系(20 μL)包含正、反向引物(10.0 μmol·L-1)各1 μL,cDNA模板DNA 1 μL,SYBR Green Dye 10 μL,DEPC水7 μL。qRT-PCR反应在ABI 7500荧光定量PCR仪(ABI公司,美国)上进行,反应条件:95℃预变性1 min,95℃变性15 s,60℃延伸30 s,共40个循环,最后72℃延伸45 s。利用2-ΔΔCt法对上述基因的相对表达量进行计算。
2 结果
2.1 数据质控与评估
在实验室条件下对意蜂工蜂进行人工饲养(图1),Am7和Am10中肠样品的测序得到raw reads平均分别为134 802 058和147 051 470条,过滤后得到clean reads平均分别为134 166 157和146 293 288条。各样品的平均Q20和Q30分别为97.34%和93.86%(表1)。Am7与Am10的组内各生物学重复之间的Pearson相关系数均在0.97以上,说明各样品的重复性较好(图2)。上述结果说明本研究的测序数据质量良好,可用于进一步分析。
随机挑选9个lncRNA进行RT-PCR验证,电泳结果显示其中有8个lncRNA均能扩增出符合预期的目的片段,说明本研究预测出的绝大多数的lncRNA真实存在(图3)。相关引物信息详见表2。进一步对上述8个验证的lncRNA的上下游基因进行预测,其中TCONS_00020918可调控西方蜜蜂王浆主蛋白1编码基因,TCONS_00021005可调控西方蜜蜂未知蛋白LOC725PPP,TCONS_00025221可调控西方蜜蜂未知蛋白LOC726321、LOC100577788和LOC102656594。

横纵坐标均表示基因表达量(FPKM)The horizontal and vertical coordinates represent gene expression level (FPKM)

表1 RNA-seq数据概览
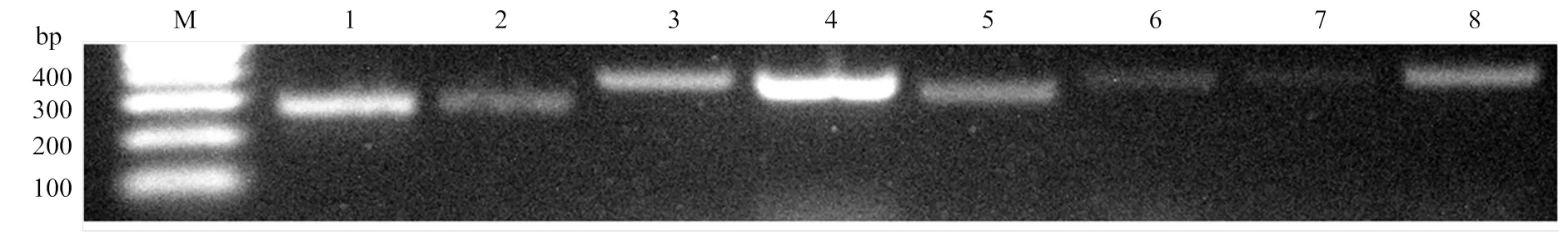
M: DNA marker; 1: TCONS_00020918; 2: TCONS_00021005; 3: TCONS_00019675; 4: TCONS_00019678; 5: TCONS_00025221; 6: TCONS_00025232; 7: TCONS_00025235; 8: TCONS_00025236

表2 RT-PCR与RT-qPCR引物信息
2.2 DElncRNA及其上下游基因分析
LncRNA的差异表达分析结果显示,在Am7 vs Am10比较组中共有3 890个DElncRNA,包括2 005个上调lncRNA和1885个下调lncRNA。lncRNA可通过影响其编码基因的上下游基因而发挥作用。共预测出1 698个DElncRNA的上下游基因,其中上游基因、下游基因和重叠基因分别为485、535和593个。对DElncRNA编码基因的上下游基因进行GO分类,结果显示它们分布在生物学进程、细胞组分和分子功能3大类,涉及42个GO term,富集基因数最多的前10个GO term分别为结合(355 genes)、细胞进程(315 genes)、代谢进程(299 genes)、单细胞进程(262 genes)、催化活性(259 genes)、细胞(147 genes)、细胞组分(147 genes)、细胞膜(138 genes)、细胞膜组分(131 genes)和细胞器(117 genes)(图4)。
进一步对DElncRNA编码基因的上下游基因进行KEGG pathway富集分析,结果显示它们富集在251个pathway,富集基因数最多的前10位pathway是Hippo信号通路(36 genes)、Wnt信号通路(19 genes)、癌症microRNA(16 genes)、癌症通路(16 genes)、剪接体(15 genes)、PI3K-AKt信号通路(14 genes)、碳代谢(12 genes)、单纯疱疹感染(12 genes)、癌症蛋白聚糖(12 genes)和神经营养因子信号通路(11 genes)(图5-A)。Hippo信号通路的概貌如图5-B所示。
2.3 DElncRNA的调控网络分析
LncRNA可以作为一种竞争性内源RNA,可通过结合miRNA减少靶向结合mRNA的miRNA数,从而减轻miRNA对mRNA的抑制作用[38]。利用软件预测DElncRNA靶向结合的miRNA,以及miRNA靶向结合的mRNA,其中靶向结合miRNA最多的DElncRNA为TCONS_00004891(36 miRNAs)、XR_001704571.1(28 miRNAs)和TCONS_00024939(27 miRNAs),靶向结合mRNA最多的miRNA为ame-miR-6001-3p(98 mRNAs)、mir-136-y(61 mRNAs)和mir-410-y(38 mRNAs)。进一步利用Cytoscape软件对DElncRNA-miRNA-mRNA调控网络进行可视化,结果显示上调、下调lncRNA与miRNA和mRNA间均形成复杂的调控网络(图6),共有125个lncRNA预测到靶向结合的miRNA,其中仅有6个lncRNA靶向结合1个miRNA,其余的119个lncRNA均靶向结合2个或更多个miRNA。
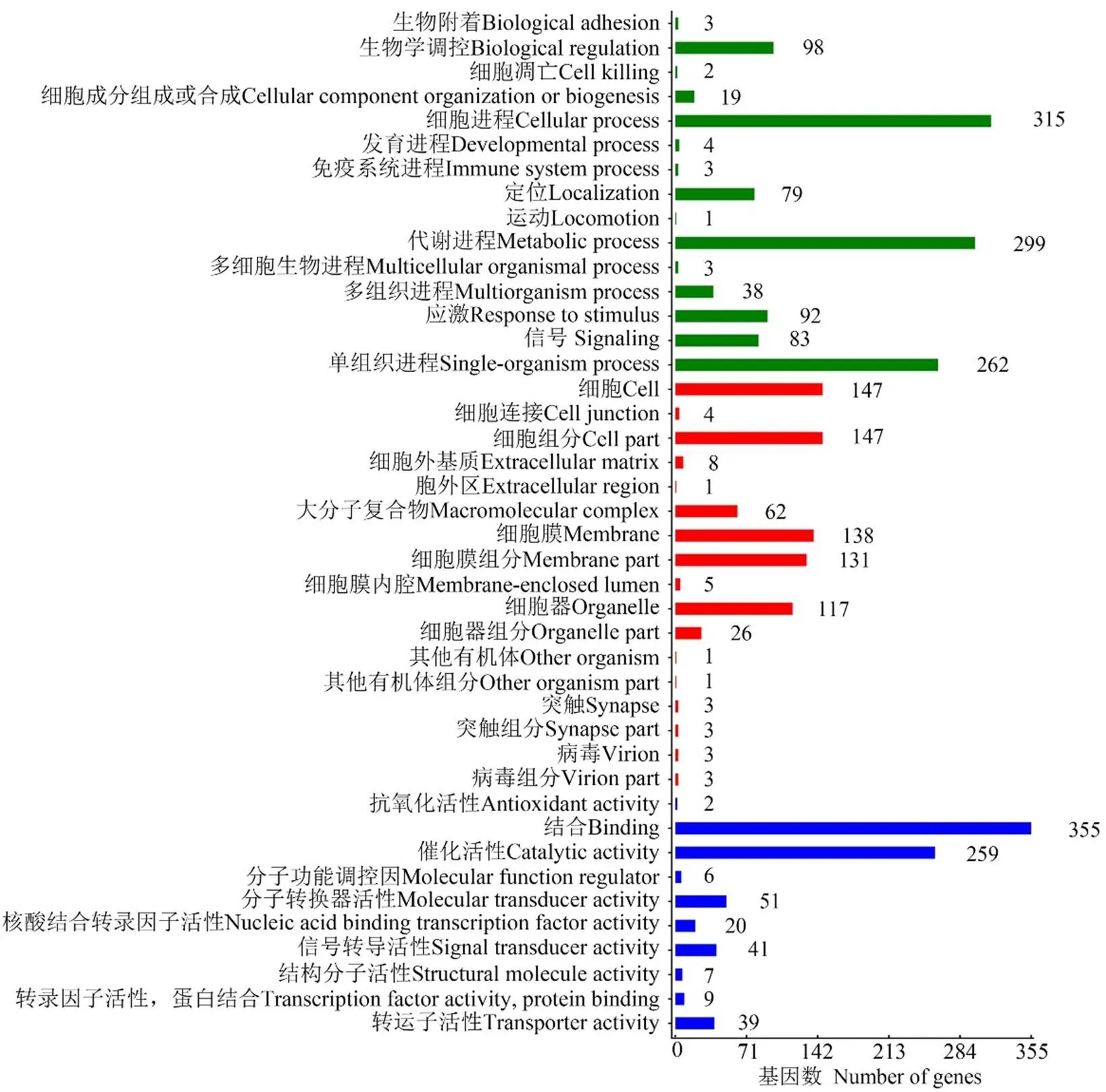
图4 DElncRNA上下游基因的GO分类
2.4 DElncRNA的qRT-PCR验证
随机挑取6个DElncRNA进行RT-qPCR验证,结果显示其中有5个DElncRNA表达水平的变化趋势和转录组数据中相应DElncRNA表达水平的变化趋势一致(图7),说明本研究中的测序数据真实可靠。相关引物序列信息详见表2。对验证的5个DElncRNA进行上下游基因分析,发现TCONS_00029692可调控肥大细胞脱粒肽编码基因,TCONS_00038012可调控未知蛋白LOC100576733编码基因。
3 讨论
此前,蜜蜂肠道的相关研究集中在肠道微生物方面,有关蜜蜂肠道的发育机理、ncRNA在肠道发育过程中作用的研究未见报道。前人研究结果表明lncRNA与生物的神经发育、细胞组织发育、细胞周期调控、干细胞分化、免疫应答和癌症发生等过程关系密切。近年来,随着二代测序技术的迅速发展和广泛应用,越来越多的lncRNA在动物、植物和微生物中被鉴定出来,但蜜蜂lncRNA的研究较为滞后,相关信息极为有限。为探究意蜂工蜂中肠的发育机理及调控机制,本研究结合RNA-seq技术和链特异性建库方法对意蜂7和10日龄工蜂中肠进行测序,基于高质量的测序数据共预测出6 353个lncRNA,RT-PCR验证的扩增成功率高达88.9%,表明本研究预测出的绝大多数lncRNA真实存在。相对于非特异性建库,特异性建库的方法在合成cDNA第二链时,将dTTP替换为dUTP,加上接头后再用UNG酶降解第二链,因而该方法可以确定转录本来自正链或负链,故能更准确地获得基因的结构和基因表达信息。lncRNA的表达具有时间和组织特异性,本研究的测序对象是意蜂工蜂中肠组织,它们在其他组织中是否表达以及表达水平有待于进一步研究。本研究中人工饲养工蜂的过程中仅饲喂糖水,工蜂的肠道内会有少量流体且多聚集在后肠,为排除杂质对测序结果的影响,舍弃了后肠,将中肠组织用于测序。测得的原始数据包含工蜂本身的数据和少量的中肠内微生物的数据,为排除后者的干扰,对原始数据进行了严格的过滤和质控,先比对核糖体数据库以去除rRNA数据,再比对西方蜜蜂基因组去除未比对上的数据,保留下来的能比对上的数据用于后续的生物信息学分析,考虑到昆虫和微生物的物种亲缘关系较远,二者的基因保守性很低,因而比对上参考基因组的数据理论上皆为意蜂工蜂中肠本身的数据。

A:Pathway富集分析enrichment analysis;B:Hippo信号通路概貌General picture of Hippo signaling pathway

A:上调lncRNA的lncRNA-miRNA-mRNA网络lncRNA-miRNA-mRNA networks of up-regulated lncRNAs;B:下调lncRNA的lncRNA-miRNA-mRNA网络 lncRNA-miRNA-mRNA networks of down-regulated lncRNAs

A: XR_409562.2; B: TCONS_00011956; C: TCONS_00012589; D: TCONS_00038012; E: TCONS_00029692
LncRNA的功能与其编码基因坐标临近的蛋白编码基因相关,位于上游的lncRNA可能与启动子或共表达基因的其他顺式作用元件有交集,从而在转录或转录后水平对基因的表达进行调控[39]。本研究中,DElncRNA的上下游基因达到1 697个,GO富集分析结果显示分别有299个和4个上下游基因涉及代谢进程和发育进程,有259个上下游基因涉及氧化磷酸化,表明DElncRNA参与意蜂工蜂中肠的物质和能量代谢过程调控。本研究发现,315个上下游基因涉及细胞进程,表明DElncRNA广泛参与意蜂工蜂中肠的细胞生命活动。此外,还发现分别有92个和3个上下游基因涉及应激反应和免疫系统进程,暗示相应的DElncRNA在意蜂工蜂中肠发育中发挥调控作用。王浆主蛋白(MRJP)为幼虫发育提供必要的可利用氮元素,在蜜蜂行为中有潜在的调控作用,还参与决定蜂群中雌性蜂的级型分化等,其中MRJP1在蜂王浆主蛋白中含量最丰富,所占比例达31%,占蜂王浆中水溶性蛋白的48%[40]。本研究发现,TCONS_00020918可调控西方蜜蜂MRJP1编码基因,表明该lncRNA可能在意蜂的营养吸收、级型分化中发挥重要的调控功能。此外,多个DElncRNA可能调控未知蛋白编码基因,具体调控关系的明确有赖于Nr数据库蛋白功能注释信息的完善及进一步的实验验证。
蜜蜂肠道是物质和能量合成与代谢的主要场所。本研究中,DElncRNA上下游基因的KEGG富集分析结果显示,共有221个上下游基因富集在多达65个物质代谢通路上,包括甘油酯代谢(5 genes)和脂肪酸的生物合成(2 genes)等10条脂代谢相关通路,柠檬酸盐循环(6 genes)和丙酮酸盐代谢(2 genes)等13条碳水化合物代谢相关通路,赖氨酸降解(3 genes)和精氨酸生物合成(2 genes)等13条氨基酸代谢相关通路,以及嘌呤代谢(10 genes)和嘧啶代谢(1 gene),表明DElncRNA广泛参与意蜂工蜂中肠发育过程中的物质代谢调控。物质合成与代谢往往伴随着旺盛的能量代谢,本研究发现共有15个上下游基因富集在甲烷代谢(5 genes)、氧化磷酸化(5 genes)和硫代谢(2 genes)等能量代谢通路,表明相应的DElncRNA在意蜂工蜂中肠的能量代谢方面具有重要的调控功能。蜜蜂肠道是物质消化吸收的主要位置。本研究发现28个上下游基因富集在7个消化系统通路上,包括胰腺分泌(8 genes)、蛋白消化和吸收(2 genes)、胃酸分泌(7 genes)、脂肪体消化和吸收(4 genes)、唾液分泌(4 genes)、维生素消化和吸收(1 genes)和胆汁分泌(2 genes),表明DElncRNA参与调控意蜂工蜂中肠对物质的消化和吸收。Hippo信号通路可以调节器官的大小[41-42],也能与其他信号通路相互作用共同调节中肠组织的稳态[43]。Notch信号影响细胞正常形态发生的多个过程,包括多能细胞分化、细胞凋亡、细胞增殖及细胞边界的形成[44]。本研究发现,分别有36、19和8个上下游基因分别富集在Hippo、Wnt和Notch信号通路。此外,还发现7、3、3和7个上下游基因富集在TGF-beta、mTOR、Hedgehog和胰岛素信号通路。结果表明相应的DElncRNA可通过参与调控上述信号通路对意蜂工蜂中肠发育进行调控。
角质层和围食膜是昆虫免疫防御的第一道防线[45]。如果病原微生物突破第一道防线,包括蜜蜂在内的昆虫将会启动细胞和体液免疫抵抗病原的入侵。蜜蜂的细胞免疫包括内吞作用、黑化作用、吞噬作用、蛋白的酶促水解等,体液免疫主要为抗菌肽的合成与释放[46]。本研究中,DElncRNA上下游基因的KEGG pathway富集分析结果显示分别有11、9、7、5和4个上下游基因富集在溶酶体、内吞作用、泛素蛋白水解、吞噬体和黑化作用等细胞免疫通路,此外,分别有13、3、1和1个上下游基因富集在MAPK、Jak-STAT、NF-Kappa B和Toll-like受体等体液免疫通路。上述结果表明相应的DElncRNA同时参与了宿主的细胞和体液免疫的调控过程,推测其在宿主的免疫防御过程发挥关键作用。
Salmena等提出内源性竞争RNA(ceRNA)假说[38],该假说认为lncRNA和mRNA等RNA可以通过miRNA应答原件(MRE)与miRNA竞争性结合,从而对基因表达进行调控。此后,ceRNA假说已被越来越多的研究[47]所证明。为进一步揭示DElncRNA的作用,本研究通过靶向关系预测DElncRNA结合的miRNA以及miRNA结合的mRNA,并构建三者间的调控网络,发现部分上调与下调lncRNA位于调控网络中心位置且结合较多的miRNA。其中,部分lncRNA可结合多个miRNA,如TCONS_00004891、XR_001704571.1和TCONS_00024939分别结合36、28和27个miRNA,表明这些lncRNA可能在意蜂工蜂中肠发育过程中发挥重要的调控功能。此外,也有部分lncRNA共同靶向结合一个miRNA,如有多达50个lncRNA能够靶向结合ame-miR-6001-3p。COLLINS等在研究真社会昆虫等级分化过程中发现,bte-miR-6001-3p在欧洲熊蜂()等级分化的幼虫中差异表达显著,ame-miR-6001-3p在西方蜜蜂等级分化的幼虫中差异表达较弱,说明miR-6001-3p影响蜜蜂卵巢发育[48]。上述结果表明DElncRNA可作为ceRNA吸附结合miRNA,从而对意蜂工蜂中肠发育过程进行调控。目前,lncRNA的功能研究多集中在人类、哺乳动物和重要疾病领域[17],在果蝇中也有少量研究[49],但包括蜜蜂[18-21]在内的绝大多数昆虫的lncRNA研究仍处于初级阶段,相关的lncRNA信息十分有限。本研究预测出的多数lncRNA,其具体的调控关系尚不明确,需要进一步克隆出lncRNA全长序列并深入探究其功能,这是未来的研究方向。
本研究仅对意蜂7和10日龄工蜂中肠进行了测序和lncRNA相关分析,若要全面解析意蜂工蜂中肠的发育机理及调控机制,需要对更多日龄的工蜂中肠进行测序,进而在全局水平进行更加深入的分析,例如对所有DElncRNA进行基因权重共表达分析(WGCNA)。
4 结论
通过对意蜂工蜂中肠发育过程中的lncRNA及其功能进行深入分析,提供了意蜂工蜂中肠发育过程中的DElncRNA信息,揭示了DElncRNA广泛参与意蜂工蜂中肠的新陈代谢、细胞活动和免疫调控并作为竞争性内源RNA(ceRNA)发挥作用,为关键lncRNA的筛选和功能研究提供了必要的数据支持。
[1] PARK D, JUNG J W, CHIO B S, JAYAKODI M, LEE J, LIM J, YU Y, CHOI Y S, LEE M L, PARK Y, CHOI I Y, YANG T Y, EDWARDS O R, NAH G, KWON H W. Uncovering the novel characteristics of Asian honey bee,by whole genome sequencing., 2015, 16(1): 1.
[2] National Research Council, Division on Earth and Life Studies, Board on Agriculture and Natural Resources, Board on Life Sciences. Committee on the Status of Pollinators in North America.. Washington, D.C: National Academies Press, 2007.
[3] 周冰峰. 蜜蜂饲养管理学. 厦门: 厦门大学出版社, 2002.
ZHOU B F.. Xiamen: Xiamen University Publishing Company, 2002. (in Chinese)
[4] DJEBALI S, DAVIS C A, MERKEL A, DOBIN A, LASSMANN T, MORTAZAVI A, TANZER A, LAGARDE J, LIN W, SCHLESINGER F, XUE C, MARINOV G K, KHATUN J, WILLIAMS B A, ZALESKI C, ROZOWSKY J, RODER M, KOKOCINSKI F, ABDELHAMID R F, ALIOTO T, ANTOSHECHKIN I, BAER M T, BAR N S, BATUT P, BELL K, BELL I, CHAKRABORTTY S, CHEN X, CHRAST J, CURADO J, DERRIEN T, DRENKOW J, DUMAIS E, DUMAIS J, DUTTAGUPTA R, FALCONNET E, FASTUCAM, FEJES-TOTH K, FERREIRA P, FOISSAC S, FULLWOOD M J, GAO H, GONZALEZ D, GORDON A, GUNAWARDENA H, HOWALD C, JHA S, JOHNSON R, KAPRANOV P, KING B, KINGSWOOD C, LUO O J, PARK E, PERSAUD K, PREALL J B, RIBECA P, RISK B, ROBYR D, SAMMETH M, SCHAFFER L, SEE L H, SHAHAB A, SKANCKE J, SUZUKI A M, TAKAHASHI H, TILGNER H, TROUT D, WALTERS N, WANG H, WROBEL J, YU Y, RUAN X, HAYASHIZAKI Y, HARROW J, GERSTEIN M, HUBBARD T, REYMOND A, ANTONARAKIS S E, HANNON G, GIDDINGS M C, RUAN Y, WOLD B, CARNINCI P, GUIGO R, GINGERAS T R.Landscape of transcription in human cells., 2012, 489(7414): 101-108.
[5] GORODKIN J, HOFACKER I L. From structure prediction to genomic screens for novel non-coding RNAs., 2011, 7(8): e1002100.
[6] KANDURI C. Kcnq1ot1: a chromatin regulatory RNA., 2011, 22(4): 343-350.
[7] TRIPATHI V, SHEN Z, CHAKRABORTY A, GIRI S, FREIER S M, WU X, ZHANG Y, GOROSPE M, PRASANTH S G, LAL A, PRASANTH K V. Long noncoding RNA MALAT1 controls cell cycle progression by regulating the expression of oncogenic transcription factor B-MYB., 2013, 9(3): e1003368.
[8] LI M, SUN X, CAI H, SUN Y, PLATH M, LI C, LAN X, LEI C, LIN F, BAI Y, CHEN H. Long non-coding RNA ADNCR suppresses adipogenic differentiation by targeting miR-204., 2016, 1859(7): 871-882.
[9] ULITSKY I, BARTEL D P. LincRNAs: genomics, evolution, and mechanisms., 2013, 154(1): 26-46.
[10] HUNG T, WANG Y, LIN M F, KOEGEL A K, KOTAKE Y, GRANT G D, HORLINGS H M, SHAH N, UMBRICHT C, WANG P, WANG Y, KONG B, LANEROD A, BORRESEN-DALE A L, KIM S K, VAN D V M, SUKUMAR S, WHITFIELD M L, KELLIS M, XIONG Y, WONG D J, CHANG H Y. Extensive and coordinated transcription of noncoding RNAs within cell cycle promoters., 2011, 43(7): 621-629.
[11] YOON J H, ABDELMOHSEN K, SRIKANTAN S, YANG X, MARTINDALE J L, DE S, HUARTE M, BECKER K G, GOROSPE M. LincRNA-p21 suppresses target mRNA translation., 2012, 47(4): 648-655.
[12] CHOONIEDASS-KOTHARI S, EMBERLEY E, HAMEDANI M K, TROUP S, WANG X, CZOSNEK A, HUBE F, MUTAWE M, WATSON P H, LEYGUE E. The steroid receptor RNA activator is the first functional RNA encoding a protein., 2004, 566(1/3): 43-47.
[13] OGAWA Y, SUN B K, LEE J T. Intersection of the RNA interference and X-inactivation pathways., 2008, 320(5881): 1336-1341.
[14] KENIRY A, OXLEY D, MONNIER P, KYBA M, DANDOLO L, SMITS G, REIK W. The H19 lincRNA is a developmental reservoir of miR-675 which suppresses growth and Igf1r., 2012, 14(7): 659-665.
[15] MUDGE J M, HARROW J. Creating reference gene annotation for the mouse C57BL6/J genome assembly., 2015, 26(9/10): 366-378.
[16] HON C C, RAMILOWSKI J A, HARSHBARGER J, BERTIN N, GOUGH J, DENISENKO E, SCHMEIER S, POULSEN T M, SEVERIN J, LIZIO M, KAWAJI H, KASUKAWA T, ITOH M, BURROUGHS A M, NOMA S, DJEBALI S, ALAM T, MEDVEDEVA Y A, TESTA A C, LIPOVICH L, YIP C W, ABUGESSAISA I, MENDEZ M, HASEGAWA A, TANG D, LASSMANN T, HEUTINK P, BABINA M, WELLS C A, KOJIMA S, NAKAMURA Y, SUZUKI H, DAUB C O, DE-HOON M J, ARNER E, HAYASHIZAKI Y, CARNINCI P, FORREST A R. An atlas of human long non-coding RNAs with accurate 5′ ends., 2017, 543(7644): 199-204.
[17] 朱斌, 梁沛, 高希武. 长链非编码RNA (lncRNA)及其在昆虫学研究中的进展. 昆虫学报, 2016, 59(11): 1272-1281.
ZHU B, LIANG P, GAO X W. Long noncoding RNAs (lncRNAs) and their research advances in entomology., 2016, 59(11): 1272-1281. (in Chinese)
[18] 郭昱, 苏松坤, 陈盛禄, 张少吾, 陈润生. LncRNA在蜜蜂级型分化中的功能研究. 生物化学与生物物理进展, 2015, 42(8): 750-757.
GUO Y, SU S K, CHEN C L, ZHANG S W, CHEN R S. The function of lncRNAs in the caste determination of the honeybee., 2015, 42(8): 750-757. (in Chinese)
[19] HUMANN F C, TIBERIO G J, HARTFELDER K. Sequence and expression characteristics of long noncoding RNAs in honey bee caste development–potential novel regulators for transgressive ovary size., 2013, 8(10): e78915.
[20] CHEN X, MA C, CHEN C, LU Q, SHI W, LIU Z G, WANG H H, GUO H K. Integration of lncRNA-miRNA-mRNA reveals novel insights into oviposition regulation in honey bees., 2017, 5: e3881.
[21] JAYAKODI M, JUNG J W, PARK D, AHN Y J, LEE S C, SHIN S Y, CHIN C,YANG T J, KWON H W. Genome-wide characterization of long intergenic non-coding RNAs (lincRNAs) provides new insight into viral diseases in honey beesand., 2015, 16(1): 680.
[22] BABENDREIER D, JOLLER D, ROMEIS J, BIGLER F, WIDMER F. Bacterial community structures in honeybee intestines and their response to two insecticidal proteins., 2007, 59(3): 600-610.
[23] KOCH H, ABROL D P, LI J, SCHMID-HEMPEL P. Diversity and evolutionary patterns of bacterial gut associates of corbiculate bees., 2013, 22(7): 2028-2044.
[24] ELLEGAARD K M, TAMARIT D, JAVELIND E, OLOFSSON T C, ANDERSSON S G, VASQUEZ A. Extensive intra-phylotype diversity in lactobacilli and bifidobacteria from the honeybee gut., 2015, 16(1): 284.
[25] MOHR K I, TEBBE C C. Diversity and phylotype consistency of bacteria in the guts of three bee species (Apoidea) at an oilseed rape field., 2006, 8(2): 258-272.
[26] ENGEL P, MARTINSON V G, MORAN N A. Functional diversity within the simple gut microbiota of the honey bee., 2012, 109(27): 11002-11007.
[27] GILLIAM M, VALENTINE D K. Enterobacteriaceae isolated from foraging worker honey bees,., 1974, 23(1): 38-41.
[28] LANGMEND B, TRAPNELL C, POP M, SALZBERG S L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome., 2009, 10(3): R25.
[29] KIM D, PERTEA G, TRAPNELL C, PIMENTEL H, KELLEY R, SALZBERG S L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions., 2013, 14(4): R36.
[30] Honeybee Genome Sequencing Consortium.Insights into social insects from the genome of the honeybee., 2006, 443(7114): 931-949.
[31] KONG L, ZHANG Y, YE Z Q, LIU X Q, ZHAO S Q, WEI L, GAO G. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine., 2007, 35(Web Server issue): 345-349.
[32] SUN L, LUO H, BU D, ZHAO G, YU K, ZHANG C, LIU Y, CHEN R, ZHAO Y.Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts., 2013, 41(17): e166.
[33] ROBINSON M D, MCCARTHY D J, SMYTH G K. EdgeR: a bioconductor package for differential expression analysis of digital gene expression data., 2010, 26(1): 139-140.
[34] REHMSMEIER M, STEFFEN P, HOCHSMANN M, GIEGERICH R. Fast and effective prediction of microRNA/target duplexes., 2004, 10(10): 1507-1517.
[35] BETEL D, WILSON M, GABOW A, MARKS D S, SANDER C. The microRNA.org resource: targets and expression., 2008, 36(Database issue): 149-153.
[36] ALLEN E, XIE Z, GUSTAFSON A M, CARRINGTON J C. MicroRNA-directed phasing during trans-acting siRNA biogenesis in plants., 2005, 121(2): 207-221.
[37] SMOOT M E, ONO K, RUSCHEINSKI J, WANG P L, IDEKER T. Cytoscape 2.8: new features for data integration and network visualization., 2011, 27(3): 431-432.
[38] SALMENA L, POLISENO L, TAY Y KATS L, PANDOLFI P P. Ahypothesis: The Rosetta stone of a hidden RNA language?, 2011, 146(3): 353-358.
[39] FLIRIAN K, JOSHUA T M. Functional classification and experimental dissection of long noncoding RNAs., 2018, 172(3): 393-407.
[40] 赵亚周, 田文礼, 胡熠凡, 彭文君. 蜜蜂蜂王浆主蛋白 (MRJPs)的研究进展. 应用昆虫学报, 2012, 49(5): 1345-1353.
ZHAO Y Z, TIAN W L, HU Y F, PENG W J. Research advances in major royal jelly proteins of honeybee., 2012, 49(5): 1345-1353. (in Chinese)
[41] HALDER G, JOHNSON R L. Hippo signaling: growth control and beyond., 2011, 138(1): 9-22.
[42] PAN D. The hippo signaling pathway in development and cancer., 2010, 19(4): 491-505.
[43] CAMARGO F D, GOKHALE S, JOHNNIDIS J B, FU D, BELL G W, JAENISCH R, BRUMMELKAMP T R. YAP1 increases organ size and expands undifferentiated progenitor cells., 2007, 17(23): 2054-2060.
[44] BITEAU B, HOCHMUTH C E, JASPER H.JNK activity in somatic stem cells causes loss of tissue homeostasis in the aginggut., 2008, 3(4): 442-455.
[45] ORIHEL T C. The peritrophic membrane: its role as a barrier to infection of the arthropod host//. Academic Press, 1975: 65-73.
[46] ARONSTEIN K A, MURRAY K D. Chalkbrood disease in honey bees., 2010, 103(Suppl. 1): 20-29.
[47] KARRENTH F A, TAY Y, PERNA D, ALA U, TAN S M, RUST A G, DENICOLA G, WEBSTER K A, WEISS D, PEREZ-MANCERA P A, KRAUTHAMMER M, HALABAN R, PROVERO P, ADAMS D J, TUVESON D A, PANDOLFI P P.identification of tumor suppressive PTEN ceRNAs in an oncogenic BRAF-induced mouse model of melanoma., 2011, 147(2): 382-395.
[48] COLLINS D H, MOHORIANU I, BECKERS M, MOULTON V, DALMAY T, BOURKE A F. MicroRNAs associated with caste determination and differentiation in a primitively eusocial insect., 2017, 7: 45674.
[49] Li E H, Zhao X, Zhang C, LIU W. Fragile X mental retardation protein participates in non-coding RNA pathways., 2018, 40(2): 87-94.
(责任编辑 岳梅)
Differential expression analysis of long non-coding RNAs during the developmental process ofworker’s Midgut
GUO Rui1, GENG Sihai1, XIONG Cuiling1, ZHENG Yanzhen1, FU Zhongmin1, WANG Haipeng1, DU Yu1, TONG Xinyu1, ZHAO Hongxia2, CHEN Dafu1
(1College of Bee Science, Fujian Agriculture and Forestry University, Fuzhou 350002;2Guangdong Institute of Applied Biological Resources, Guangzhou 510260)
【Objective】Long non-coding RNA (lncRNA) plays an important role in regulation of gene expression, epigenetics and cell cycle in eukaryotes. The objective of this study is to investigate the expression profile and role of lncRNAs in the developmental process ofworker’s midgut. 【Method】In this study, 7- and 10-day-old worker’s midguts of(Am7, Am10) were sequenced using RNA-seq technology and strand-specific library construction method. using Perl script, raw reads were filtered to obtain clean reads with high-quality. Bowtie tool was used to compare clean reads to the ribosome database, and TopHat2 software was employed to compare unmapped clean reads to the reference genome. CPC and CNCI softwares were utilized to predict coding capacity of the transcripts. RT-PCR was performed to identify partial lncRNAs. Investigation of differentially expressed lncRNAs (DElncRNAs) was carried out with edgeR, followed by prediction of upstream and downstream genes, for which GO and KEGG pathway enrichment analyses were performed. RNAhybrid, Miranda and TargetScan softwares were utilized together to predict target miRNAs of DElncRNAs and target genes of miRNAs, and DElncRNAs-miRNAs-mRNAs regulation networks were visualized via Cytoscape. Finally, RT-qPCR was conducted to verify reliability of the sequencing data.【Result】134 802 058 and 147 051 470 raw reads were gained from deep sequencing of Am7 and Am10, respectively, and after stringent filtration, 134 166 157 and 146 293 288 were obtained. In total, 6 353 lncRNAs were predicted, and 3 890 DElncRNAs were obtained based on expression calculation, including 2 005 up-regulated lncRNAs and 1 885 down-regulated lncRNAs. The result of RT-PCR suggested the expected signal bands could be amplified from 8 lncRNAs, implying their true existence. There were 1 793 upstream and downstream genes of DElncRNAs, which were involved in 42 GO terms, including metabolic processes, developmental processes, cellular processes, stress responses, immune system processes and so forth. They were also associated with 251 KEGG pathways, including material metabolism pathways such as carbon metabolism, purine metabolism and fatty acid biosynthesis; energy metabolism pathways such as sulfur metabolism, methane metabolism and oxidative phosphorylation; signaling pathways such as Hippo, Wnt and Notch signaling pathways; cellular immune pathways such as lysosome, endocytosis and ubiquitin mediated proteolysis; humoral immune pathways such as MAPK, Jak-STAT and NF-kappa B pathways, these results demonstrated the DElncRNAs were involved in the material and energy metabolism, cell life activity and immunity regulation in the developmental process ofworker’s midgut. Further analysis showed TCONS_00020918 might play a regulatory part in the nutrient absorption and caste differentiation in the worker’s midgut.Analysis of regulation networks demonstrated that complex networks existed between DElncRNAs and target miRNAs and mRNAs, partial DElncRNAs lie in the central of the networks and link many miRNAs, and partial miRNAs could be bound by many DElncRNAs, which indicated that these DElncRNAs might play an important role during the developmental process of the worker’s midgut. Finally, 5 DElncRNAs were randomly selected for RT-qPCR assay, and the result proved the reliability of sequencing data in this study.【Conclusion】DElncRNA is widely involved in the metabolism, cellular activity and immune regulation ofworker’s midgut, and plays a role as a competitive endogenous RNA (ceRNA). The results provide the necessary data support for the screening and functional study of key lncRNA.
; midgut; development; long non-coding RNA; upstream and downstream genes
10.3864/j.issn.0578-1752.2018.18.016
2018-03-17;
2018-05-08
国家自然科学基金(31702190)、国家现代农业产业技术体系建设专项资金(CARS-44-KXJ7)、福建省教育厅中青年教师教育科研项目(JAT170158)、福建农林大学科技创新专项基金(CXZX2017343)、福建省大学生创新创业训练计划(201610389053)
郭睿,E-mail:ruiguo@fafu.edu.cn。耿四海,E-mail:15737313592@163.com。郭睿和耿四海为同等贡献作者。通信作者陈大福,E-mail:dfchen826@fafu.edu.cn
