气—质联用法同时检测高脂食品中14种光引发剂
2018-10-10徐文泱
徐文泱 陈 华
(湖南省食品质量监督检验研究院,湖南 长沙 410017)
光引发剂是紫外光印刷油墨中非常重要的组成部分,它受紫外光照射后,吸收光的能量,分裂成2个活性自由基,引发光敏树脂和活性稀释剂发生连锁聚合,使胶黏剂交联固化,作为油墨固化的催化剂被广泛用于紫外光油墨的印刷包装上[1]。常见的小分子光引发剂主要分为水性均裂碎片型光引发剂和水性氢转移型光引发剂。前者有水性安息香衍生物类(如安息香、安息香双甲醚、安息香乙醚、安息香异丙醚、安息香丁醚)、苯乙酮衍生物(如α,α-二乙氧基苯乙酮、α-羟烷基苯酮、α-胺烷基苯酮、二苯基乙酮、α,α-二甲氧基-α-苯基苯乙酮)以及烷基苯酮类,水性氢转移型光引发剂的代表化学结构为蒽醌、硫杂蒽酮以及二苯甲酮,其中水性二苯甲酮类又可按其在二苯甲酮母核引入的基团分为阳离子、阴离子和非离子类型[2]。光引发剂因其合成较为简单,品种多样,价格低廉而备受市场的青睐。
但由于光引发剂是溶脂性小分子物质,极易从包装材料迁移到食品中,造成食品污染[3]。已有报道证实食品中也能检测出二苯甲酮、异丙基硫杂蒽酮等光引发剂[4]。试验[5-6]表明光引发剂苯甲酮可增加罹患肿瘤、尿道下裂的风险,引发多种过敏性皮肤病。因此,食品中多种光引发剂检测方法的建立可以为此类污染物的风险监测提供技术支持,提高食品质量安全监管能力,健全预警应急机制。
目前关于光引发剂的检测方法以食品包装材料方向居多[7-9]。刘艳等[10]建立了塑料及纸塑类包装材料中9种光引发剂的气相色谱质谱法。在食品中检测光引发剂的方法研究相对较少,主要集中在乳制品及饮料中光引发剂的检测[11-13]上。前处理方法主要有固相萃取、固相微萃取技术和QuEChERS方法等,使用的仪器多为气相色谱—质谱联用仪和液相色谱串联质谱仪。张耀海等[11]研究了8种光引发剂在橙汁、苹果汁、桃汁、菠萝汁和凉茶5种食品中的GC/MS-MS检测方法,结果表明,方法的回收率偏差较大,最低为60.4%,最高达到99.1%,结果的相对偏差最高接近16%。另有文献[12]采用GC-MS法对乳制品中的异丙基硫杂蒽酮检测方法进行了报道,检出限在0.007 mg/kg左右。
由于光引发剂多为脂溶性小分子物质,其向高脂高蛋白的食品中迁移概率更高。本研究拟选取14种有代表性的酮、醚、酯类光引发剂,拓展了研究食品种类,针对肉类、乳制品、糕点等高油富含蛋白质的食品,着力于建立一种灵敏度高、选择性好,回收率和精密度等技术参数均能达到方法学指标要求的高通量GC-MS检测方法,以同时检测出高脂食品中的14种光引发剂,为研究此类光引发剂的迁移规律提供依据。
1 材料与方法
1.1 材料与试剂
2-氯噻吨酮、苯甲酰甲酸甲酯、2-苯甲酰苯甲酸甲酯、2-甲基二苯甲酮、4-氯-二苯甲酮、四乙基米氏酮、2-羟基-甲基苯丙酮、2,4-二乙基硫杂蒽-9-酮、3甲基-二苯甲酮、米氏酮、4-甲基二苯甲酮、安息香二甲醚、二苯甲酮、对二甲氨基苯甲酸异辛酯:99%,德国Dr Ehrenstorfer公司;
丙酮、正己烷、乙酸乙酯、石油醚、甲醇:色谱纯,上海安谱实验科技股份有限公司;
HLB小柱:60 mg,3 mL,上海安谱实验科技股份有限公司;
PSA试剂管:2 mL(150 mg MgSO4,50 mg PSA,50 mg GCB,50 mg C18),上海安谱实验科技股份有限公司;
试验用食品:市售。
1.2 仪器与设备
气相色谱—质谱联用仪:Agilent 7890A-5975C MSD型,美国安捷伦科技公司;
高速冷冻离心机:Neofuge 1600R型,力康生物医疗科技控股有限公司;
分析天平:SPS401F型,奥豪斯仪器(上海)有限公司;
涡混振荡仪:CM-1000型,东京理化器械株式会社。
1.3 方法
1.3.1 色谱条件 色谱柱:Agilent 19091S-433(30 m×250 μm×0.25 μm);进样口温度:250 ℃;柱流速:1 mL/min。进样量:1 μL;程序升温条件:初始温度为60 ℃保持1 min,以20 ℃/min升至180 ℃保持3 min,以5 ℃/min升至280 ℃保持5 min。
1.3.2 质谱条件 EI 源。离子源温度为230 ℃,四极杆温度150 ℃,分别采用SCAN模式和SIM模式,在整个确认试验中,14种光引发剂的色/质谱图信息见表1。针对检测的光引发剂品种多,保留时间相对集中的特点,在选择扫描离子时考虑以下几个原则:① 尽量选择丰度高的碎片离子;② 选择的监测离子扣除背景后,其信噪比应尽量大于3。
当试样待测液保留时间和目标化合物一致(±0.5%),其质谱碎片离子质荷比吻合,丰度比与标准品的偏差符合表2,则可对试样待测液中的光引发剂进行定性确认。
1.3.3 标准溶液的配制 准确称量14种标样各0.1 g(精确至0.000 1 g)于10 mL容量瓶中,稀释溶剂为丙酮,各光引发剂母液浓度为10 g/L。分别移取0.1 mL母液于100 mL容量瓶中,以丙酮定容至刻度,此为标准储备液,各光引发剂的浓度均为10 μg/mL。系列工作溶液现配现用。
1.3.4 样品前处理 将样品匀质后,准确称取2~5 g(精确至0.000 1 g),以20 mL丙酮—正己烷(体积比2∶8)提取,涡混振荡15 min后,于4 000 r/min离心5 min,取上清液加入到活化后的HLB小柱,以10 mL丙酮—正己烷进行洗脱,收集洗脱液,浓缩至近干以丙酮定容到2 mL,移取1 mL到PSA净化管中,离心后上机。

表1 14种光引发剂的保留时间和质谱中的选择离子及丰度比Table 1 Retention time and selected ion of 14 photoinitiators
表2气相色谱—质谱定性确证相对离子丰度最大容许误差
Table 2 The maximum allowable error of relative ion abundance by gas chromatography-mass spectrometry %

相对丰度GC-MS相对离子丰度最大允许误差(50,∞)±10(20,50]±15(10,20]±20[0,10]±50
2 结果与讨论
2.1 色谱条件的优化
试验考察了2种不同的色谱程序升温条件:
(1) 60 ℃保持1 min,以20 ℃/min到150 ℃保持1 min,再以2 ℃/min 升到270 ℃保持8 min。
(2) 60 ℃保持 1 min,以 20 ℃/min 到 240 ℃ 保持0 min,再以2 ℃/min 到 270 ℃保持25 min。
从图1、2可以看到,在第(1)种程序升温条件下14种光引发剂仅出现了9个峰,且苯甲酰甲酸甲酯和2-羟基-甲基苯丙酮的2峰分离度差,在第(2)种程序升温条件下14种光引发剂均能出峰,但峰与峰过于紧密,影响定量分析。因此将第(2)种程序升温条件进行改进。将出峰较多的前段升温程序放缓,即当60 ℃保持1 min后,以10 ℃/min 到120 ℃保持2 min,再以5 ℃/min 到240 ℃保持5 min,以2 ℃/min升温至270 ℃保持15 min。由图3可以看到,14种光引发剂均能检测出,并且峰形良好,分离度较佳。
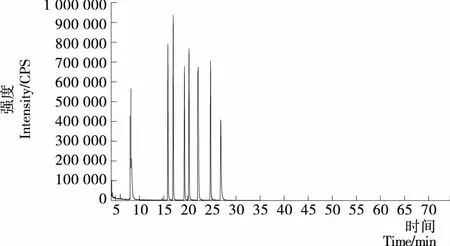
图1 程序升温条件(1)的总离子流图
图1 GC-MS chromatogram of the mixture of 14 photoinitiators with 1stcolumn temperature programme
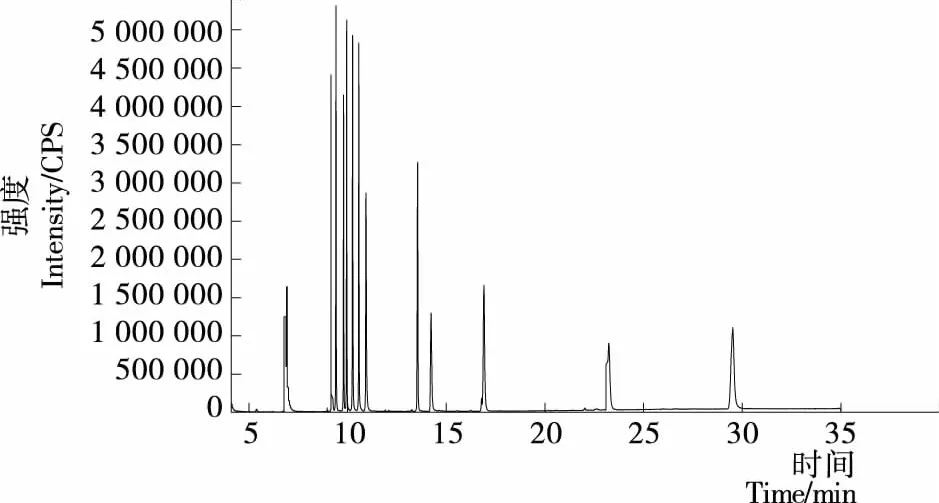
图2 程序升温条件(2)的总离子流图
图2 GC-MS chromatogram of the mixture of 14 photoinitiators with 2ndcolumn temperature programme
14种光引发剂均为脂溶性化合物,而高脂高蛋白食品种类繁多,基质相对复杂。而采用气/液相质谱联用仪定量分析复杂基质中化合物含量时应考虑基质效应。将空白样品经过前处理的提取净化以及浓缩至近干后,分别加入相同体积不同浓度的标准溶液,振荡离心后即得相应质量浓度的基质标准工作溶液。按式(1)对基质效应的程度进行评估。当ME≥20%时,需考虑基质效应对结果准确度带来的影响。由表3可知,ME均小于20%,在定量分析中无需考虑基质效应。可以看到本试验前处理条件下测得的光引发剂检出限低于文献[14]报道,对目标物的痕量检测具备一定优势。
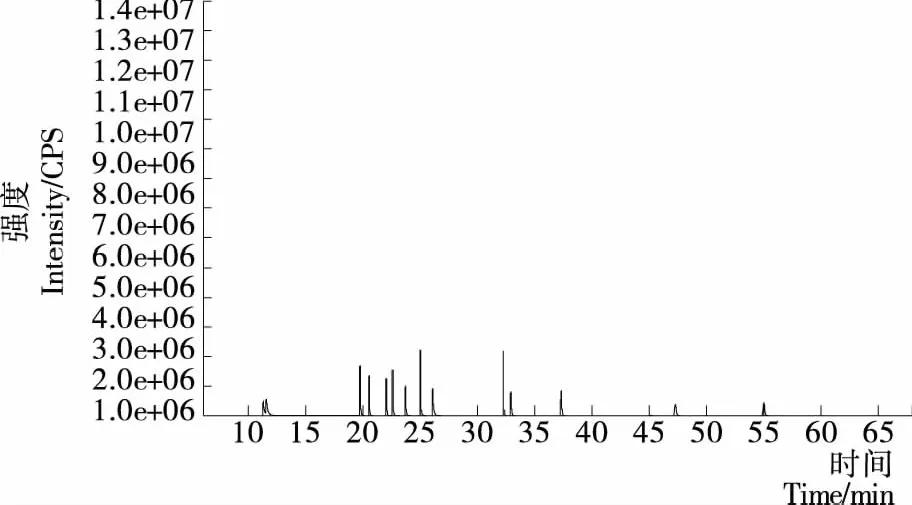
图3 程序升温条件(3)的总离子流图
图3 GC-MS chromatogram of the mixture of 14 photoinitiators with 3rdcolumn temperature programme
(1)
式中:
ME——基质效应,%;
M1——基质匹配校准曲线斜率;
M2——纯溶剂标准曲线斜率。
2.2 提取条件的确认
选取的14种光引发剂酮类、酯类结构居多,并有少量硫醚类物质。根据文献[15-17]及其化学性质,选取甲醇、乙酸乙酯、丙酮以及乙腈为参考提取溶剂。在配制标准溶液过程中发现,2-氯噻吨酮不溶于甲醇和乙腈,改选用极性稍弱的丙酮和乙酸乙酯。由于乙酸乙酯的出峰时间早于苯甲酰甲酸甲酯的出峰时间,导致了它可能被乙酸乙酯的溶剂峰覆盖,且硫醚类物质在乙酸乙酯溶液中不稳定。因此,初步选择丙酮为试验的提取溶剂。由于高脂食品易发生乳化现象,在用有机溶剂萃取前先加入少量氯化钠以达到破乳的目的。已有研究[14]表明,非极性溶剂和中等极性溶剂的结合使用有利于提高目标提取物的稳定性,并且有效地促进目标物和杂质的分离。因此,选取体积比1∶1的丙酮—正己烷、丙酮—石油醚和丙酮—二氯甲烷作为提取溶剂进行回收率试验。提取和净化操作按1.3.4进行,图4为不同的提取溶剂与回收率关系示意图。
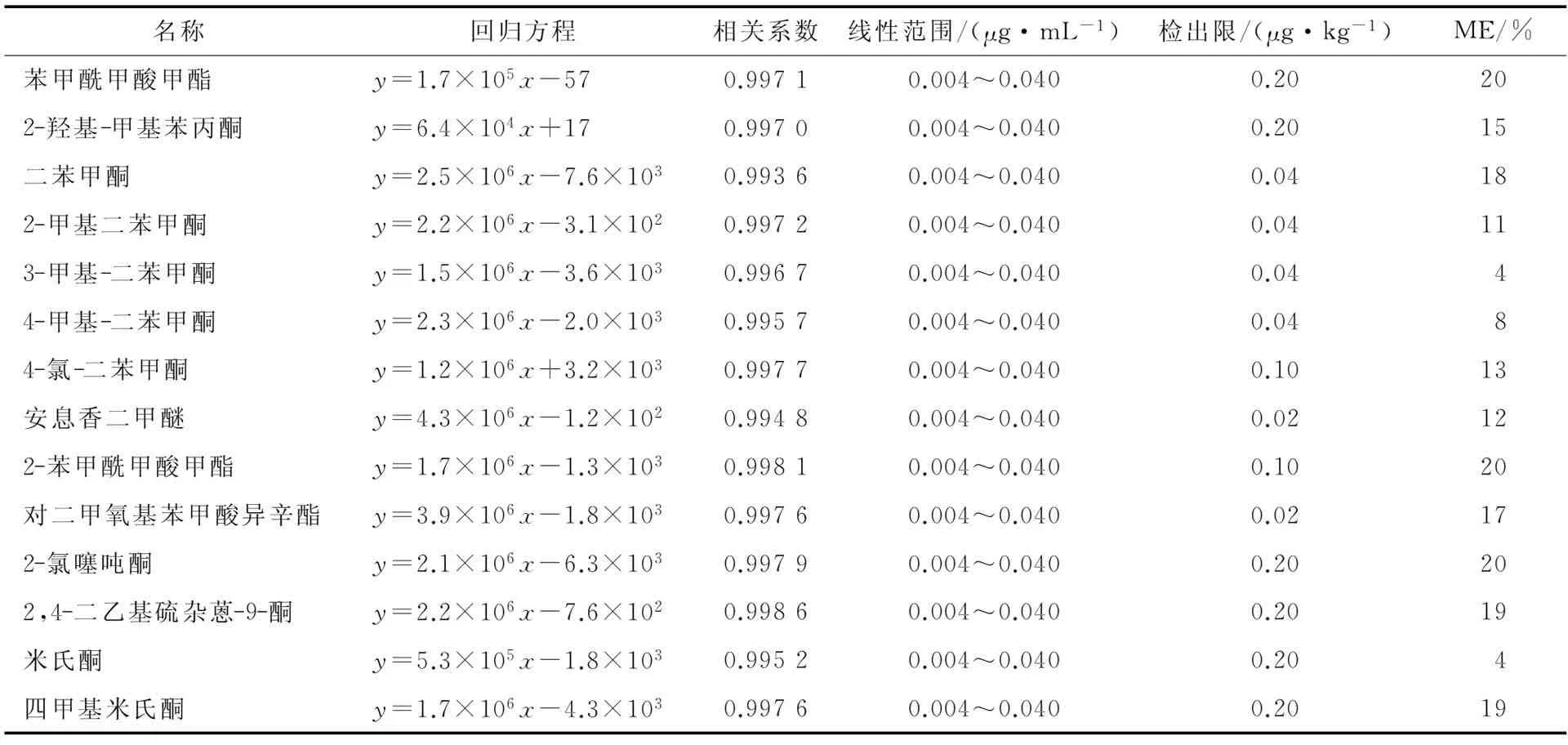
表3 14种光引发剂的回归方程、相关系数、线性范围及检出限Table 3 Linear equation, correlation coefficients, linearity range and LOD of 14 photoinitiators
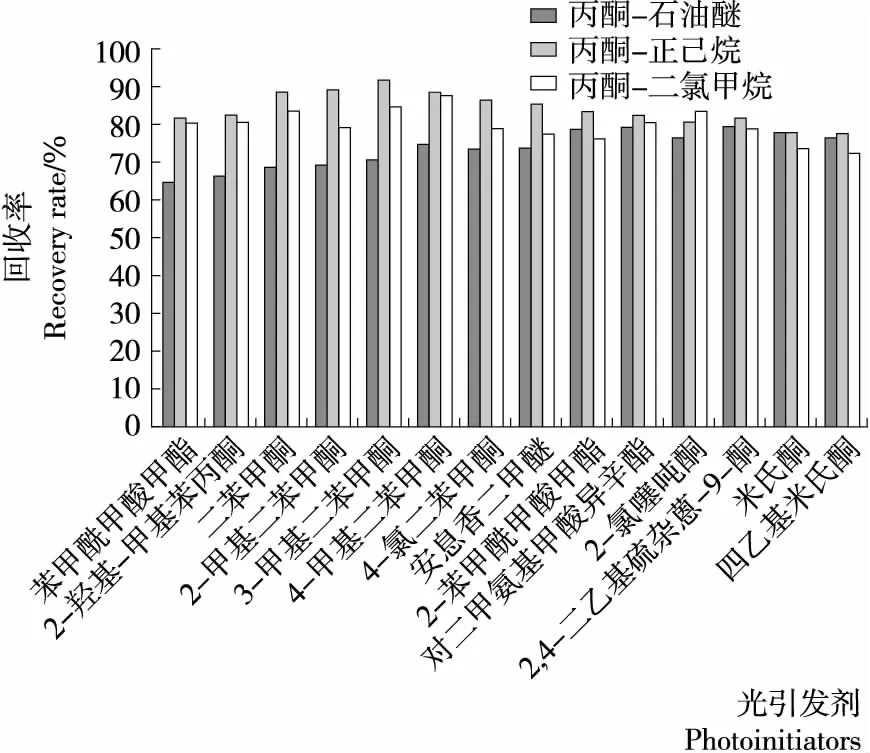
图4 不同的提取溶剂对回收率的影响Figure 4 Effects of different solvents on the recoveries
由图4可知,丙酮—正己烷对14种光引发剂的平均回收率最高。进一步建立正交试验法考察萃取条件。分别选取提取溶剂的体积比、萃取温度、提取时间作为萃取条件的3个影响因素,设计了三因素三水平正交试验。试验设计见表4。试验结果和极差分析见表5。
结果表明,丙酮的比例减少有利于回收率的提高,可能是提取效率较高,在提取了目标物的同时杂质也较易溶在此溶剂中。由表5可知,最佳提取条件为丙酮和正己烷体积比2∶8,萃取温度20 ℃,提取时间15 min。按该条件进行3次平行验证实验,光引发剂的平均回收率为93.1%,优于其他试验条件所得的回收率,因此确认提取条件为以丙酮—正己烷(体积比2∶8)为提取溶剂,20 ℃下提取15 min即可最大效率地提取出所有目标化合物。
2.3 净化条件的优化
2.3.1 吸附剂的选择 QuEChERS方法是分散固相萃取技术的衍生和发展,其核心成分是N-丙基乙二胺吸附剂。目前,在食品及包装材料中光引发剂检测的报道中用到此类净化方法的较多[11,18]。由于本试验中食品基质较为复杂,采用单一的QuEChERS方法进行净化可能无法取得较为理想的净化效果。因此,尝试采用HLB小柱和QuEChERS技术相结合的模式进行净化,以达到去除基质干扰,降低检出限的目的。由图5可以看出,单采用QuEChERS方法净化在加标浓度较低时的回收率大部分均低于采用两者相结合的方法进行净化的。HLB柱中的吸附剂是亲水亲脂平衡聚合物,它的作用基团是苯基、乙烯基和吡咯烷酮基,应用机理是通过非极性的相互作用来保留目标物。在其净化的基础上用PSA进行下一步净化以去除碳水化合物、脂肪酸、有机酸、酚类和色素,可有利于提高净化效果,去除基质的干扰,也能提高方法灵敏度,达到痕量分析的目的。图5的回收率也可显示将这两种净化吸附剂结合起来使用效果最优。

表4 正交试验因素水平表Table 4 Factors and levels of orthogonal experiment
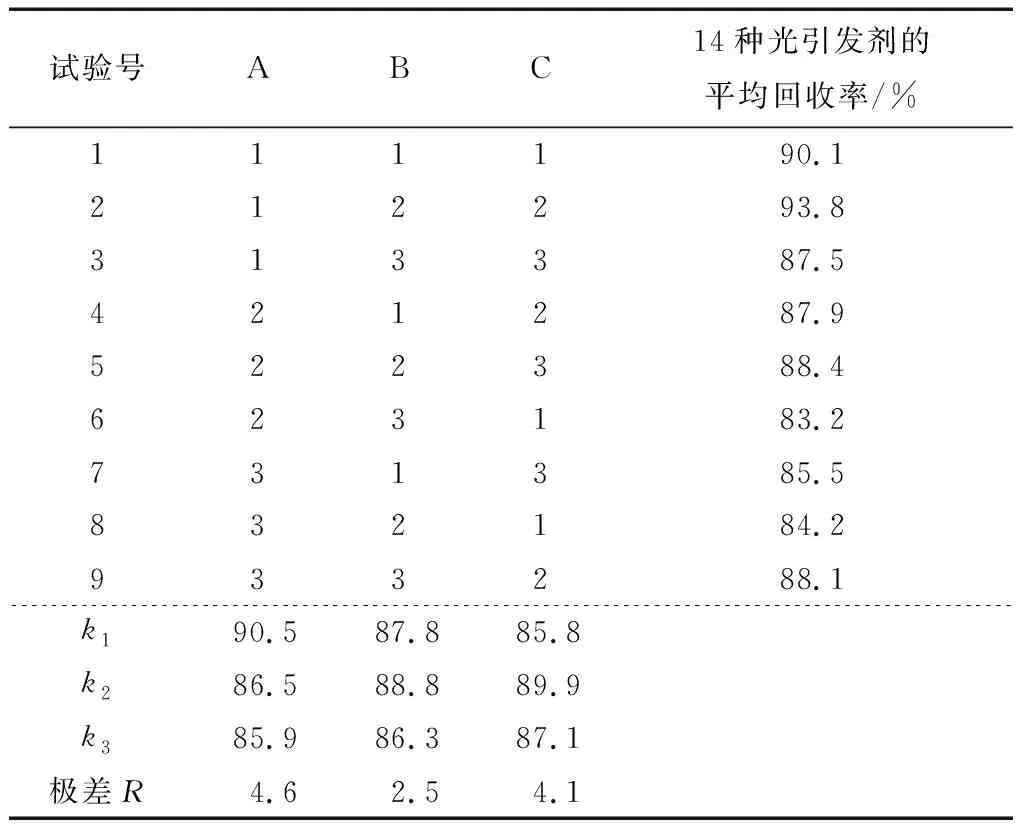
表5 正交试验结果与极差分析Table 5 Orthogonal experiment result
2.3.2 过柱体积 由于固相萃取柱容积的局限性,需要在尽可能对样品体积进行浓缩的前提下保证萃取效率。因此,试验对由丙酮—正己烷提取后的上清液在进行HLB过柱净化时的溶液体积也进行了考察。分别选用15,30,50 mL待净化溶液过柱,考察不同体积对待测化合物回收率的影响。当过柱体积为30 mL时,所有的待测物回收率处于3种水平中的最高值。当溶液为50 mL时,所有的待测物回收率均有所下降。而当待净化溶液为15 mL时,米氏酮和四甲基米氏酮的回收率相比30 mL时的下降最为明显,达到了30%以上。
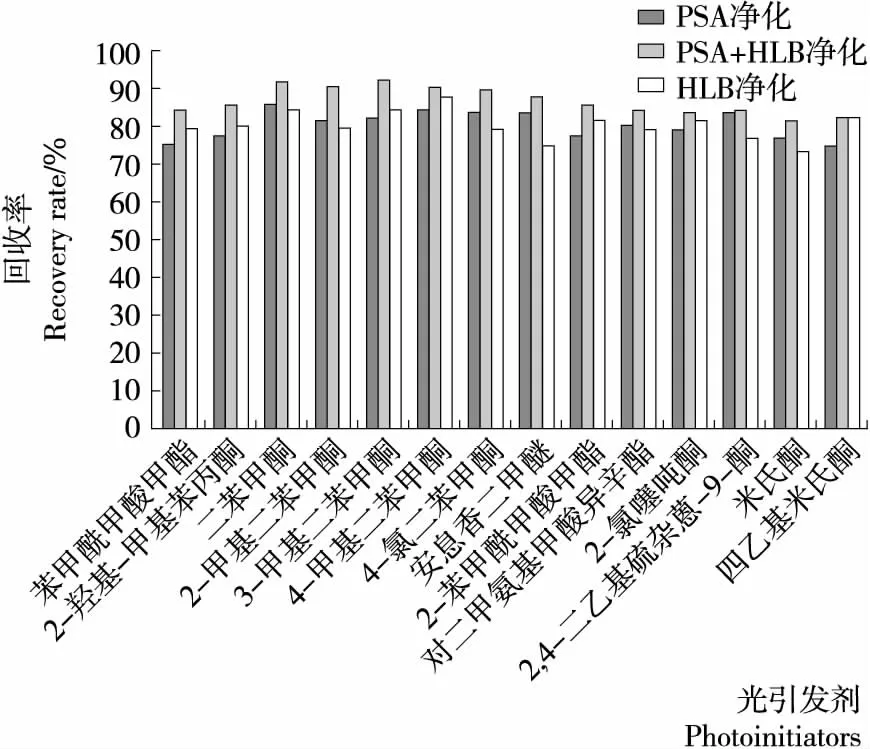
图5 3种净化方法对样品中14种光引发剂加标回收率的影响
图5 Effects of three purification methods on the recoveries of 14 photoinitiators in the spiked samples
2.4 回收率和精密度
为了验证本方法的准确度和精密度,选取了3个有代表性的浓度做加标试验,进行6次平行样测试,方法的回收率和精密度见表6。由表6可知,RSD均<10%,在3个浓度下的加标回收率均满足GB/T 27404—2008 对方法确认技术参数的要求。本试验中14种光引发剂的回收率为73.0%~92.4%,回收率的相对标准偏差为4.1%~8.9%。

表6 14种光引发剂的加标回收率及精密度Table 6 The recoveries and RSD of 14 photoinitators
2.5 实际样品检测
采用本方法对40个样品(代表基质为酸奶、牛奶、腊肉和面包)进行了检测,共有2个牛奶样品分别检测出了二苯甲酮和2-氯噻吨酮,其他样品中均未检出光引发剂。
3 结论
本试验建立了气相色谱—质谱联用法同时测定高脂高蛋白食品中14种光引发剂的方法。方法采用HLB固相萃取小柱和PSA净化管进行联合净化。所得的方法检出限为0.02~0.20 μg/kg,14种光引发剂的回收率为73.0%~92.4%,回收率的相对标准偏差均小于10%,方法的检出限低,重现性好,准确度较高。从方法前处理到检测结果的确认少于3 h,实现了14种光引发剂在此类食品中的快速高效检测。下一步将围绕食品类别与光引发剂检出及含量关系进行研究,并扩大食品类别及光引发剂的种类对方法的适用性进行进一步评估。
