附加染色体异常的t(8;21)阳性儿童急性髓细胞白血病临床分析*
2018-09-14姚新原赵利师安曦洲温贤浩郭玉霞管贤敏胡艳妮肖剑文重庆医科大学附属儿童医院血液肿瘤科儿科学重庆市重点实验室儿童发育疾病研究省部共建教育部重点实验室重庆400014
姚新原,赵利师,安曦洲,于 洁,宪 莹,温贤浩,郭玉霞,管贤敏,胡艳妮,肖剑文(重庆医科大学附属儿童医院血液肿瘤科/儿科学重庆市重点实验室/儿童发育疾病研究省部共建教育部重点实验室,重庆400014)
急性髓细胞白血病(AML)是一种异质性较强的血液系统恶性肿瘤,50%以上的AML患儿伴克隆性染色体异常。按照2008年世界卫生组织(WHO)造血与淋巴组织肿瘤分类,该类患儿属于伴重现性遗传学异常的AML。伴t(8;21)(q22;q22)的AML[即t(8;21)-AML]可占AML的20%~40%[1-2],t(8;21)-AML患儿通常对化疗反应较好,无事件生存期(EFS)长,临床上归入低危组。t(8;21)-AML患儿常伴其他重现性染色体改变(即附加染色体异常),而附加染色体异常对t(8;21)-AML患者临床及预后影响的研究较少[3-6]。本研究回顾性分析t(8;21)-AML患儿的临床资料并探讨附加染色体异常对其临床特征及预后的影响,现报道如下。
1 资料与方法
1.1 一般资料 选取2008年1月至2015年12月重庆医科大学附属儿童医院血液肿瘤科收治的初诊AML患儿268例,以75例t(8;21)-AML患儿为研究对象,其中男45例,女30例。所有患儿均参照2008年WHO造血与淋巴组织肿瘤分类制定的标准完善骨髓细胞学、免疫分型、细胞及分子遗传学检查。继发性AML、婴儿AML、未完善上述检查及外院接受过化疗者未纳入本研究。本研究得到重庆医科大学附属儿童医院医学伦理委员会批准,患儿家属均签署知情同意书。
1.2 方法
1.2.1 细胞形态学 骨髓涂片瑞氏染色并结合组织化学染色,镜下分类计数至少200个细胞,按照FAB分型进行细胞形态学检查并分为M0~M7亚型[7]。
1.2.2 免疫分型 治疗前采集肝素抗凝骨髓2~3 mL分离单个核细胞,采用活细胞间接荧光标记和流式细胞仪检测。所用单抗包括淋系的 CD2、CD3、CD5、CD7、CD10、CD19、CD20 及 CD22,髓 系 的 CD13、CD14、CD15、CD33 及 CD117,巨核系的 CD41、CD61及干系的CD34。阳性标准:淋系及髓系抗原阳性细胞大于20%,CD34阳性细胞大于10%。
1.2.3 染色体核型分析 采集治疗前乙二胺四乙酸(EDTA)抗凝骨髓2~4 mL行24~48 h短期培养,G显带技术对大于或等于20个处于分裂中期的细胞核型进行核型分析,参照ISCN2009标准描述[8],核型异常大于或等于2条染色体或3个断裂点为复杂核型。
1.2.4 RUNX1/RUNX1T1融合基因检测 治疗前取肝素抗凝骨髓2~3 mL,逆转录聚合酶链反应(RT-PCR)技术检测AML1/ETO融合基因[9]。
1.2.5 治疗方案及疗效评价 参照儿童AML诊疗建议化疗[10],疗效标准参照文献[10-11]的标准进行评估,随访时间截至2016年5月。随访指标包括完全缓解(CR)率及EFS。EFS定义为从确诊到第1次事件(包括复发、死亡)时间或末次随访时间。
1.3 统计学处理 采用SPSS20.0统计软件对数据进行统计学处理。计量资料以表示,组间比较采用独立样本t检验或单因素方差分析。计数资料以例数或率表示,组间比较采用χ2检验。生存分析应用Kaplan-Meier法,组间比较采用log-rank检验。以P<0.05为差异有统计学意义。
2 结 果
2.1 染色体核型分析 75例患儿中50例(66.67%)伴附加染色体异常,其中性染色体缺失38例(占所有患儿比例50.67%,占附加染色体异常者比例为76.00%),包括-Y 25例,-X 13例(12例女性,1例男性)。5例患儿同时还伴附加其他染色体异常。性染色体缺失外,其他附加染色体异常以常染色体长臂部分缺失为主,包括8例 9q-、2 例 7q-,1 例 5q-和 1 例 6q-。14 例患儿存在复杂核型,其中6例伴性染色体缺失。8例伴附加染色体异常患儿因例数太少未纳入分析,其余67例患儿分为3组:仅有t(8;21)组(A组,25例)、t(8;21)仅伴性染色体缺失组(B组,28例)和复杂核型组(C组,14例)。
2.2 临床资料 75例患儿发病年龄(96.31±36.23)个月。初诊血常规:白细胞计数(WBC)为(15.78±15.93)×109L−1,血红蛋白(Hb)为(66.70±19.21)g/L,血小板计数(Plt)为(38.71±34.45)×109L−1。乳酸脱氢酶(LDH)为(711.47±661.56)U/L。56例行全身骨骼 X 线片检查,17例异常(30.36%)。3组患儿中19例未做骨片检查。纳入研究的3组患儿发病年龄、性别构成、初诊WBC、Hb、LDH和骨片异常比例比较,差异均无统计学意义(P>0.05),但A组患儿Plt水平较B、C组低,差异均有统计学意义(P<0.05),见表1。
2.3 细胞形态学分型及RUNX1/RUNX1T1基因分析 75例患儿AML-M2型63例(84.00%),AML-M5型8例(10.67%),AML-M4型 4例(5.33%)。在纳入 3组 67例中,AML-M2型在3组中的比例分别为96.00%、82.14%和85.71%,三者比较,差异无统计学意义(P>0.05)。75例均检测出RUNX1/RUNX1T1基因,染色体核型与基因检测结果相符。见表2。

表1 各组患儿临床及实验室特征比较

表2 各组患儿FAB亚型的分布情况及比较(n)
2.4 疗效分析 放弃及CR后接受造血干细胞移植患儿均未纳入疗效分析,40例(A、B、C组分别为18例、16例和6例)根据儿童AML诊疗建议方案化疗[10]。1个DAE疗程后33例CR、7例未CR,CR率为82.50%。未CR患儿中1例接受第2疗程DAE治疗并达CR,6例放弃。CR患儿中2例放弃,31例继续化疗,6例复发,A组因经济原因放弃和感染死亡各1例。3组患儿第一疗程CR率比较,差异无统计学意义(P=0.526)。3组患儿平均 EFS 分别为(56.446±8.790)个月、(75.844±8.176)个月和(45.000±7.361)个月,3组比较,差异均无统计学意义(P=0.670)。见图 1。
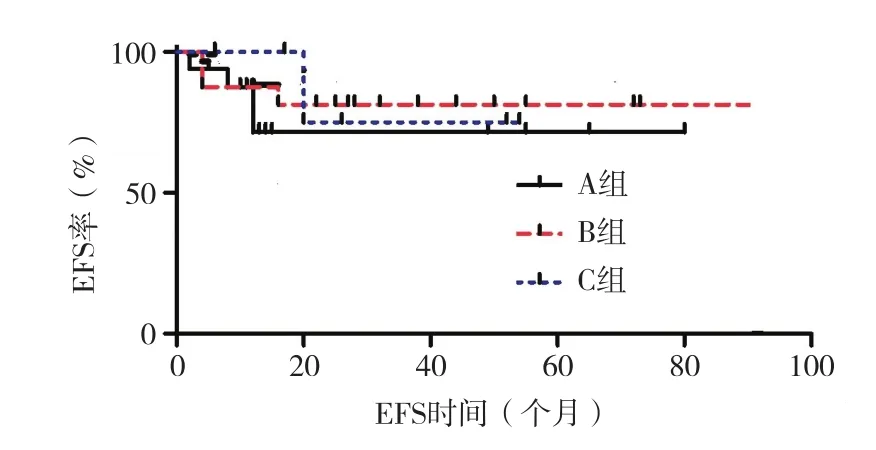
图1 3组患儿无事件生存曲线
3 讨 论
AML是一组临床及遗传学上均具异质性的恶性疾病,占儿童白血病的15%~25%[12]。50%~93%的AML患者有细胞遗传学异常,而细胞遗传学改变已成为指导AML诊断治疗的重要标志[12-14]。t(8;21)是AML中最常见的染色体重排,该易位导致21q22的RUNX1基因易位至8q22并与该位置上RUNX1T1基因并置形成RUNX1/RUNX1T1基因(又称AML/ETO基因),该融合基因的蛋白产物可抑制若干造血转录因子表达并引起细胞恶性分化,但其被归为AML预后良好的指标,对化疗反应好,患者使用大剂量阿糖胞苷治疗通常能获得较高CR率与长期EFS。
t(8;21)-AML常伴附加染色体异常,主要包括-Y、-X、9q-、+4、+8、-7/7q-等,其中性染色体缺失最为常见[15]。有学者报道 t(8;21)-AML中复杂核型发生率为 9%~23%。本研究结果显示,附加染色体异常检出率为66.67%,按发生率递减依次为性染色体缺失、9q-和7q-等,复杂核型发生率为18.67%,均与文献结果相似。牧启田等[16]指出,成人仅t(8;21)和伴性染色体缺失患者细胞学分型主要见于AML-M2型,其他附加染色体异常的患者则更易见于非AML-M2型。本研究结果显示,t(8;21)-AML患儿均以AML-M2型最多见且与附加染色体异常无关,提示儿童与成人存在一定差异性。本研究结果显示,仅伴性染色体缺失的 t(8;21)-AML患儿PLT水平高于单纯t(8;21)-AML组,而其他特征如发病年龄、WBC及Hb水平等相似,提示附加染色体异常可能对儿童t(8;21)-AML患者临床特点没有影响。
文献[17-19]提示,伴附加染色体异常是 t(8;21)-AML的预后不良因素,特别是9q-和性染色体缺失是t(8;21)-AML预后的不良因素。一般认为,复杂核型是AML独立的不良预后因素[20]。而本研究结果则显示,性染色体缺失、复杂核型等附加染色体异常对t(8;21)-AML患儿生存并无不良影响,其可能与儿童特点及本研究样本量较小有关。
综上所述,t(8;21)-AML患儿容易合并附加染色体异常,附加染色体异常以性染色缺失最为常见。与成人t(8;21)-AML不同的是,附加染色体异常可能不影响儿童期t(8;21)-AML的临床及实验室特征,其对t(8;21)-AML患儿生存也可能无不良影响。但本研究为单中心且样本量较少,需多中心研究附加染色体异常对t(8;21)-AML 意义。
