纳米多晶金刚石微结构及塑性形变机制的研究进展*
2018-09-14何艮春刘春梅
何艮春, 许 超, 刘春梅
(武汉科技大学 理学院, 武汉 430081)
纳米多晶金刚石是指晶粒尺寸小于100 nm的金刚石晶粒直接结合而形成的金刚石多晶材料。实验研究显示,纳米多晶金刚石不仅硬度超过了金刚石单晶,其断裂韧性、耐磨性和热稳定性也优于金刚石单晶及传统金刚石烧结体[1-8]。因此,纳米多晶金刚石不仅可以用于传统金刚石材料几乎所有的应用领域,而且已经在远红外相机ZnS菲涅尔透镜、碳化钨硬质合金等脆硬材料超精细加工中也表现出优异的性能,具有极大的应用价值[9-11]。
制备纳米尺度金刚石常见的方法有动高压法(爆轰或冲击法)[12]、化学气相沉积(CVD)法[13-14]、静高压高温无助剂烧结法[3]以及静高压高温直接转变法等[1-2, 4]。动高压法的压力和温度持续时间很短,虽利于形成纳米尺度的金刚石,但无法制备烧结完好的大尺度块体样品,常用于制备纳米金刚石粉体。化学气相沉积法虽可制备大尺度纳米金刚石材料,但由于样品的晶粒存在明显的取向,且晶粒尺寸偏大,晶粒间结合弱,导致该方法合成的纳米多晶金刚石材料力学性能只与传统金刚石相当,甚至更低。静高压高温无助剂烧结法是利用纳米金刚石粉体为初始原料,直接烧结制备纳米金刚石烧结体。实验研究显示,纳米金刚石粉体初始原料中包含的各种杂质成分无法除尽,导致烧结的样品始终包含杂质,力学性能大打折扣。目前,制备高性能纳米多晶金刚石块体最成功的方法是静高压高温直接转变法。
纳米多晶金刚石因其优异的性能和广阔的应用前景,吸引了国内外众多课题组参与研究。2003年,IRIFUNE团队在温度为2300~2500 ℃,静高压12~25 GPa压力范围内研究了高纯多晶石墨的高温高压转变行为,并首次合成了高纯透明的纳米多晶金刚石[1]。这种纳米多晶金刚石的成功合成,拉开了新型纳米超硬材料研究的序幕。2006年,IRIFUNE小组将纳米多晶金刚石的样品尺寸提升到3 mm, 2010年又将该尺寸提升到了10 mm,并对纳米多晶金刚石的实际应用性能进行了系统研究[1, 4-5, 7-11, 13-14]。DUBROVINSKAIA团队在高温静高压下直接转变C60,合成了硬度与金刚石单晶硬度相当的金刚石纳米棒聚合材料[15-16]。TANIGAKI等[17-20]利用不同晶体形态的石墨、无定形碳、炭黑、碳纳米管等,在高温静高压下直接转变合成了纳米多晶金刚石材料。
国内方面,2013年,贺端威教授课题组利用石墨等为碳源,高温静高压下直接转变合成了纳米多晶金刚石块体材料[2-3]。2014年,田永君院士团队利用纳米洋葱氮化硼为初始原料,制备了硬度超金刚石单晶的纳米孪晶立方氮化硼[21]。2015年,该团队又以纳米洋葱炭为初始原料,制备了极硬材料纳米孪晶金刚石[22]。2017年,王海阔等[23]利用石墨为初始原料,合成出直径达6 mm的纳米多晶金刚石块体材料。此外,朱品文教授团队[24]、王明智教授团队[25]、王文丹[26]等均在进行纳米多晶金刚石的相关研究。
作为极硬材料(硬度超过80 GPa的材料),纳米多晶金刚石的力学性能,特别是其塑性形变机制的研究一直以来都是大家关注的焦点。本文主要针对纳米多晶金刚石的微结构以及国内外关于纳米多晶金刚石塑性形变机制的研究及发展情况进行概述。
1 纳米多晶金刚石的微结构
1.1 高温高压直接转变石墨生成的微结构
纳米多晶金刚石优异的性能源自其微观结构。IRIFUNE小组利用晶粒尺寸小于10 μm且取向随机的石墨为原料,在压力为18 GPa、温度为2500 ℃、保温时间为10 s的条件下合成纳米多晶金刚石材料,其TEM图如图1所示[1]。实验结果显示:该纳米多晶金刚石样品中有2种不同的微结构形貌。样品部分区域金刚石晶粒形状和尺寸均匀分布,称为均匀细颗粒结构,如图1a所示;还有部分区域的金刚石晶粒呈明显的层状结构,称为非均匀层状结构,如图1b所示。以相同的石墨为原料,在压力15 GPa,2300~2500 ℃下合成的所有纳米多晶金刚石样品中均包含上述2种微观结构。其中,均匀细颗粒结构中的金刚石普遍为多面体颗粒状晶粒,且晶粒尺寸小,约为10~20 nm。图1a中的电子衍射图样显示(图1a右上角),该区域除包含立方金刚石的衍射外,不含有石墨或其他相的衍射。图1a中区域A的衍射斑沿电子反射环随机分布,形成环形衍射图案,表明样品中的金刚石颗粒都是随机取向的。另一方面,层状结构由长度为100~200 nm的层状晶体组成。层状金刚石不是直的,而是轻微弯曲的,表明层状结构不是由晶粒中的滑移变形所形成的微孪晶结构。在层状结构中,观察到了立方金刚石的对称衍射斑点(图1b右上角)。而立方金刚石(111)方向的衍射信号很强,对应于层状结构的方向,表明层状金刚石晶体平行于(111)平面。

(a)均匀细颗粒结构区域 (b)非均匀层状区域
1.2 高温高压直接转变玻璃碳和C60生成的微结构
SUMIYA等[27]做了高温高压直接转变玻璃碳和C60合成纳米多晶金刚石样品的实验。结果显示:以这2种碳源直接转变合成的纳米多晶金刚石样品只包含均匀细颗粒结构,不包含任何形式的层状结构。这些均匀细颗粒结构中的金刚石晶粒呈多面体形状,且取向随机。较低温度条件下(1600~2000 ℃)直接转变玻璃碳和C60合成的纳米多晶金刚石样品的晶粒尺寸可达10 nm以下;而温度高于2000 ℃时,晶粒尺寸会迅速长大至50~100 nm。因此,利用玻璃碳和C60在较低温度范围内(1600~2000 ℃)可以合成出晶粒极小(单个晶粒尺寸小于10 nm)且分布均匀的纳米多晶金刚石。
DUBROVINSKAIA等[28]在高温高压下直接转变C60合成了纳米多晶金刚石材料,结合透射电镜以及X射线衍射峰宽化分析显示:压力为20 GPa,温度为2300 K条件下,直接转变C60合成的纳米多晶金刚石样品的晶粒尺寸为5~12 nm。高分辨透射电镜显示:该样品的纳米金刚石晶粒拥有理想结构,不含有任何堆垛层错或者缺陷;样品中只包含极细的形状分布均匀的金刚石晶粒,不含有层状结构的金刚石颗粒。
1.3 高温高压直接转变碳纳米管生成的微结构
YUSA[20]利用金刚石对顶砧装置及激光加热技术研究了碳纳米管的高温高压直接转变行为。在17.5 GPa、2500 K的压温条件下,所得样品的TEM及电子衍射结果显示:样品的电子衍射图样不含石墨(002)晶面的最内层衍射环,而只包含金刚石的衍射环。衍射环也没有结构缺陷所对应的附加斑点衍射信号,说明纳米多晶金刚石样品扩展缺陷含量极少。TEM结果还显示:当碳纳米管在17.5 GPa、2500 K的压温条件下处理后,得到的样品不再含有碳纳米管的管状结构或洋葱结构,而是由纳米级的金刚石晶粒紧密结合的致密金刚石烧结体,晶粒尺寸在20~50 nm。该样品的晶粒尺寸分布较为均匀,未出现以石墨为原料合成的纳米多晶金刚石样品中的层状结构。
1.4 高温高压直接转变洋葱碳生成的微结构
HUANG等[22, 29-32]以洋葱碳为原料,在高温高压下合成了晶粒内包含大量孪晶的纳米多晶金刚石样品。实验显示:该样品的主要成分是立方相的金刚石,且包含大量的纳米孪晶;样品中还发现了第2种物相的存在,经选区电子衍射和X射线衍射显示,该物相与现已确定的所有碳单质的结构均不吻合,由于该物相为单斜结构,结构与金刚石十分相近,故命名为M-金刚石。TEM检测显示:M-金刚石颗粒很薄且细长,与相邻的纳米孪晶金刚石交错分布。20 GPa、2000 ℃条件下直接转变洋葱碳合成样品的TEM图片如图2所示[22]。由图2可看到:立方金刚石晶粒内部包含了高密度的层状纳米孪晶,且样品晶界无法清晰显示。纳米孪晶金刚石中的大角度晶界经常与相邻纳米晶体相交或合并,导致难以清晰地确定单个纳米金刚石颗粒。且该样品中的纳米孪晶片平均厚度低至5 nm,是迄今为止金刚石所能达到的最小尺度的微结构。
总之,不同的碳源和合成条件对纳米多晶金刚石的微观结构有重大影响。总结已有的实验结果,我们可以看到:以石墨为原料在高温高压下直接转变合成的纳米多晶金刚石样品,其微观结构具备2种特征,即存在均匀细颗粒结构和层状结构;而以玻璃碳、C60、碳纳米管以及洋葱碳等非石墨碳为初始原料合成的纳米多晶金刚石样品,其晶粒形状和尺度较为均匀,不存在片状晶粒;初始材料的微观结构对纳米多晶金刚石的微观结构有重要影响。通过选择特定微观结构的初始碳源,或是人为改变初始碳源的微观结构,先得到包含新微观结构的前驱物,然后再通过高温高压合成,可以实现纳米多晶金刚石微结构的有效调控[19, 22]。

(合成压力为20 GPa,温度为2000 ℃)(a)纳米孪晶金刚石的微观结构,插图为透明的纳米孪晶金刚石样品照片,直径1 mm;(b)沿金刚石[101]区域轴观察左图方框区域的高分辨透射电镜图片[22]。
2 纳米多晶金刚石塑性形变机制的实验研究
2.1 纳米多晶金刚石纳米压痕和维氏压痕实验研究
实验合成的纳米多晶金刚石尺寸最大只能达到10 mm左右,且其硬度极高,要使其发生塑性形变非常困难。到目前为止,测试纳米多晶金刚石材料塑性形变最方便最有效的方法是压痕法。即在一定加载力的作用下,使金刚石压头压入纳米多晶金刚石表面,产生压痕。根据压痕响应变化,来研究其塑性形变特性,即根据压痕周围微观结构的变化规律来研究纳米多晶金刚石的塑性形变机制。
SUMIYA等[8, 27, 33]用维氏硬度压头在纳米多晶金刚石表面进行压痕实验。结果显示:在压痕实验中即使加载力为4.9 N时,维氏压头也容易损坏,导致实验无法继续进行。他们又尝试了纳米压痕法,通过Berkovich压头在纳米多晶金刚石表面进行压痕实验,得到了加载力与压痕深度的关系曲线[8]。实验中,纳米压痕的最大压深接近500 nm,最大加载力不到300 mN。实验的卸载曲线显示,纳米多晶金刚石有很强的弹性回复特性。即便如此,从整个纳米压痕试验的加载力随压痕深度变化曲线可以看到,加载曲线和卸载曲线并未完全重合,从而显示样品表面发生了十分微小的塑性形变。再一次进行纳米压痕实验以确定纳米多晶金刚石的杨氏模量时,压头便发生了破碎。由此可以看出:纳米多晶金刚石拥有很高的硬度,而维氏硬度压头或者纳米压痕Berkovich压头不适用于这种材料的硬度测量。最终,用努普硬度压痕对纳米多晶金刚石材料进行了研究,初步研究了该材料在努普硬度压痕加载下塑性形变的微观机制。
2.2 纳米多晶金刚石的努普压痕实验研究
SUMIYA等[8]的努普压痕实验结果显示:仅在特定加载力范围内,才能清晰观察到压痕且不会发现裂纹。加载力大于7 N时,努普压头边缘易发生碎裂,导致压痕的形状不规则,进而无法测量。加载力小于1 N时,由于纳米多晶金刚石很强的弹性回复特性,导致无法观测到压痕。因此,选取加载力范围1~7 N,对纳米多晶金刚石进行压痕测试。实验结果显示:在加载力小于2 N时,随着加载力的降低,硬度测量值迅速上升。这是由于加载力较低时,纳米多晶金刚石材料发生弹性回复,导致压痕尺度减小所致。当加载力较大时,纳米多晶金刚石塑性形变加剧,而弹性回复减弱,因而压痕尺寸变大,硬度测量值有所降低。在压力大于15 GPa,温度高于2300 ℃条件下合成的纳米多晶金刚石样品的努普硬度均高于100 GPa。其中一些样品的硬度极高,为120~145 GPa[8]。这种高硬度纳米多晶金刚石样品的努普硬度值相当于人工合成高纯度IIa型金刚石单晶晶体(001)<100>的硬度(116~130 GPa),明显高于含0.008 8%(质量分数)氮杂质的人工合成Ib型金刚石单晶的硬度(98~106 GPa)[8]。
纳米多晶金刚石在4.9 N加载力作用下,努普硬度压痕的原子力显微镜测试结果显示:压痕周围未观察到裂纹或断裂,变形形状非常平滑,表明压痕是由塑性流动形成的[8]。而在IIa型金刚石单晶(001)面上的努普压痕周围也没有明显的裂缝,表明金刚石单晶上的压痕也主要是由塑性流动形成的。室温下,当金刚石处于特定条件时,如处于努普硬度压头或极端高压的作用下时,金刚石单晶可以发生塑性形变。因此,在4.9 N载荷下的努普压痕实验可以用来评价纳米多晶金刚石的塑性形变。
为研究纳米多晶金刚石在努普硬度压痕下的塑性形变机制,采用透射电镜对压痕周围的变形和断裂行为进行研究[27]。TEM测试结果显示:以石墨和C60为原料,高温高压直接转变合成的纳米多晶金刚石样品压痕周围区域存在裂纹。该裂纹可以分为2类[27]:处在压痕正下方的大裂纹,和在压痕周围且垂直延伸的无数纳米裂纹(100 nm或更小)。处在压痕正下方的大裂纹在压痕实验过程中并未观测到。但在使用FIB进行试样切片时,由于压痕周围的残余应力而导致相对延迟的断裂,从而形成大裂纹。纳米多晶金刚石样品的断裂韧性可以根据大裂纹的形状进行定性评价。在以石墨为原料直接转变合成的纳米多晶金刚石样品中,裂纹扩展过程因层状结构而偏转或终止。相比之下,以C60为原料合成的纳米多晶金刚石中因只有细颗粒均匀结构而没有层状结构,压痕下的裂纹会沿直线路径直接传播。以其他非石墨碳为原料合成的纳米多晶金刚石材料也表现出与后者相同的裂纹传播行为。这表明,以非石墨碳为原料合成的具有均匀细颗粒结构的纳米多晶金刚石的断裂韧性比以石墨为原料合成的具有层状结构的纳米多晶金刚石的断裂韧性要差。换言之,层状结构对提升纳米多晶金刚石的力学性能有重要的作用。
SUMIYA等[27]对压痕周围的微纳米裂纹也进行了仔细研究,其结果如图3所示。他们认为:压痕区域的形变是由于该区域材料的塑性形变和相应的微纳米裂纹所导致的。因此,通过研究这些微裂纹的行为,可以揭示纳米多晶金刚石的塑性形变机制和硬度增强的影响因素。
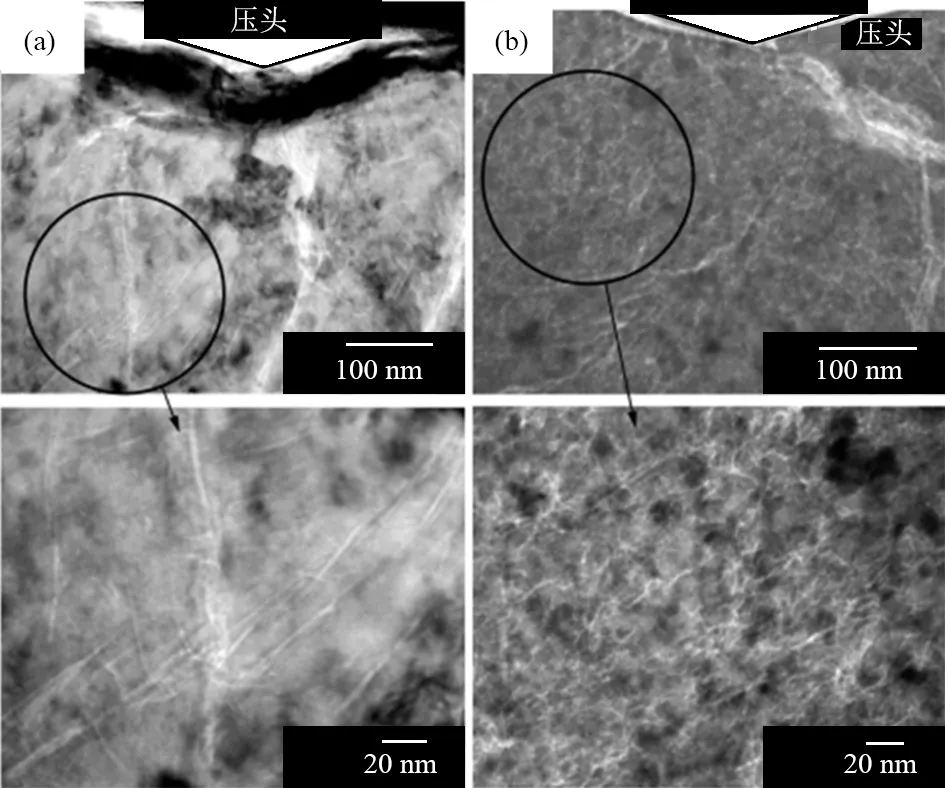
(a)15 GPa、2400℃条件下直接转变石墨合成的纳米多晶金刚石样品(努普硬度为128~138 GPa),下图为上图圆圈区域的放大图片;(b)21 GPa、1800 ℃条件下直接转变C60合成的纳米多晶金刚石样品(努普硬度为70~74 GPa),下图为上图圆圈区域的放大图片[27]。
图3a所示为以石墨为原料,在15 GPa、2400 ℃条件下直接转变石墨合成的纳米多晶金刚石样品压痕周围区域的TEM图片[27]。从图3a可以看到:该样品中的纳米裂纹传播与晶界无关,表明裂纹传播过程中,穿晶断裂占主要地位[27]。图3b是以C60为原料,在21 GPa、1800 ℃下直接转变得到的纳米多晶金刚石压痕附近区域的TEM图像[27]。从图3b可以看出:在该样品中,大部分纳米裂纹沿晶界传播,表明裂纹在该样品中传播的过程中,沿晶断裂占主导地位。以石墨为原料合成的纳米多晶金刚石材料的晶界结合力非常强(图3a),而以非石墨碳合成的纳米多晶金刚石的晶界结合力相对较弱(图3b),晶界结合的强度极大地影响了纳米多晶金刚石的塑性形变行为。实验还表明:当合成温度偏低时,如2000 ℃或更低温度下,沿晶断裂在裂纹传播过程中占主导地位[27];随着合成温度的提高,晶界结合强度也相应提高。实验结果还显示:以石墨为原料,在压力高于15 GPa、温度高于2300 ℃条件下合成的纳米多晶金刚石材料的硬度和断裂韧性最高。而以非石墨碳为原料,完全转变成金刚石所需的最低温度为1600 ℃。这意味着在较低温度范围内(2000 ℃或者更低温度)可以合成出晶粒超细(10 nm或者更低)的纳米多晶金刚石材料,但以非石墨碳为原料合成的纳米多晶金刚石的硬度和断裂韧性都不高。
根据Hall-Petch效应,细化的多晶晶粒可以导致材料的硬化,这是由于晶界附近可以实现位错的堆积,阻止位错的扩展。但是,当晶粒尺寸小到一定程度时,晶界上的位错堆积很难实现,晶界将发生滑移等现象,导致材料的软化(反Hall-Petch效应)[32]。已有的实验结果显示:非石墨碳直接转变合成的纳米多晶金刚石中,当晶粒尺寸小于10 nm时,硬度的降低不是因为晶界的滑移,而是因为晶粒与晶粒之间产生了明显的裂纹[27]。这些裂纹主要是由低温烧结过程导致的晶界结合强度不高所致。高硬度高韧性的纳米多晶金刚石材料的合成需要足够高的烧结温度(2000~2200 ℃)以促进原子的扩散,从而强化金刚石晶界的结合强度。而以石墨为原料合成的样品中出现的层状晶粒结构,可能会进一步提升纳米多晶金刚石的断裂韧性。
2.3 纳米孪晶金刚石的维氏压痕实验研究
田永君团队[22]以直径为20~50 nm的洋葱碳为原料,经高温高压直接转变合成了透明的纳米孪晶金刚石块体材料。维氏硬度和努普硬度测试均显示:该材料的硬度约为200 GPa,是金刚石单晶硬度的2倍。尽管如此,在金刚石单晶压头上施加载荷后,被测纳米孪晶金刚石样品表面依然留下永久性压痕。
图4 所示为纳米孪晶金刚石样品在9.8 N载荷下所形成压痕的原子力显微镜图片[29]。从图4可以看到:纳米孪晶金刚石表面形成了永久性的塑性形变区域。通过原子力显微镜(AFM)对压痕对角线长度进行精确测量,可较精确地测出这种极硬材料的硬度值。纳米孪晶金刚石的塑性形变也被实验证实,但遗憾的是,目前尚缺少纳米孪晶金刚石压痕周围微结构的实验数据。在纳米孪晶金刚石塑性形变机制方面,也尚未达成广泛共识。
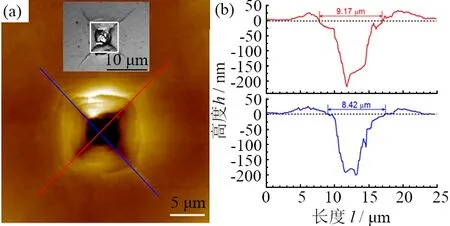
(a) AFM测量结果, 插图显示了该压痕的光学显微镜照片,通过光学显微镜测量对角线长度的位置用方框标记;(b) 对应(a)中直线的AFM轮廓线[29]。
纳米多晶金刚石的塑性形变研究除压痕法之外,前期还用纳米多晶金刚石材料制作成了硬度计压头和金刚石对顶砧等超高压实验装置[11, 34-41]。在这些实际应用中,纳米多晶金刚石也发生了弹塑性形变。但是,由于研究的侧重点不同,这些研究并未报道纳米多晶金刚石的弹塑性形变机制的实验数据。
3 纳米多晶金刚石塑性形变机制的理论研究
3.1 纳米多晶金刚石拉伸和剪切形变机制理论研究
计算机技术和材料计算相关理论的发展,使复杂结构材料的相关力学性质的计算成为可能。REMEDIAKIS等[42]构建了晶粒取向随机,粒径约为2~5 nm的纳米多晶金刚石材料模型,并利用蒙特卡罗方法模拟了力学性能与晶粒尺寸的关系。通过计算该材料的弹性常数并估算其硬度,发现该材料随晶粒尺寸的减小而发生了软化,并将软化原因归结为随着晶粒尺度的减小,晶界原子数目急剧增加。
SHA等[43]构建了超细纳米多晶金刚石材料的分子动力学模型,该模型中的原子可用中心对称参数(CSP)进行着色,来研究晶粒内部原子和晶界原子在塑性形变过程中的行为。利用分子动力学模拟研究了晶粒尺寸和温度对超细纳米多晶金刚石在拉伸载荷下的力学性能和塑性形变机制的影响,并得到了平均晶粒尺寸2.26~4.10 nm的超细纳米多晶金刚石材料的拉伸应力应变曲线。结果显示:计算所得的应力应变曲线的形状具有脆性材料的典型特征,而没有塑性形变的特征;在上述晶粒尺寸范围内(2.26~4.10 nm),断裂强度和应变对平均晶粒尺寸不敏感,平均断裂强度为113.2 GPa,对应的平均应变为0.2;不同晶粒尺寸模型对应的断裂强度和最大应变与平均值的偏差不超过3%和5%。与断裂强度和应变相反,计算的杨氏模量对平均晶粒尺寸比较敏感。杨氏模量随平均晶粒尺寸的减小而减小,表明超细纳米多晶金刚石发生了弹性软化。研究还显示:晶界原子占总原子比重随晶粒尺寸的减小上升明显,且晶界原子分数的增加与杨氏模量的减小密切相关。晶界处的原子是无序的,原子密度明显低于体密度。因此,他们认为,具有较小平均晶粒尺寸的纳米多晶金刚石样品易于变形,其杨氏模量也相应减小。
SHA等[43]还分析了超细纳米多晶金刚石材料的断裂机制,认为温度为300 K时,超细纳米多晶金刚石在拉伸载荷下的破坏过程如下:首先,晶粒沿某个具有大剪切应力的晶界滑动,在滑动过程中引起相邻3个晶粒的结点处产生微裂纹;其次,裂纹产生后会随着拉伸过程而沿应力较大的晶界传播;最后,沿晶断裂的大量产生导致样品的最终断裂。他们构建了只包含2个金刚石晶粒的模型,并进行了拉伸和剪切形变模拟计算。模拟拉伸时,拉伸应力垂直于晶界;模拟剪切时,剪切应力平行于晶界。模拟过程中,构建了3个模型,平均晶粒尺寸为4.1 nm,应变率设定为108s-1,温度保持在300 K。拉伸和剪切形变模拟所获得的应力-应变曲线结果显示:该模型的平均拉伸断裂强度为117.6 GPa(1%偏差),平均剪切断裂强度(剪切屈服应力)为53.4 GPa(2%偏差);断裂过程的可视化可以较清楚地反映断裂产生的各个阶段。
3.2 纳米孪晶金刚石维氏硬度压痕的理论研究
近期报道的晶粒内部包含高密度孪晶片的纳米孪晶金刚石材料硬度高达200 GPa,是金刚石单晶硬度值(约100 GPa)的2倍[22]。但是,这种材料的硬度增强机制不能由Hall-Petch效应来解释,因为该材料中的典型孪晶片厚度为3~6 nm,处在反Hall-Petch效应区域范围内(微结构尺寸低于10 nm)。为了阐明纳米孪晶金刚石的硬度增强机制和压痕下的形变机制,LI等[44]对该材料在维氏硬度压头加载下的结构和应力响应进行了第一性原理研究。他们构建了包含不同厚度孪晶片的材料模型(如图5所示)。
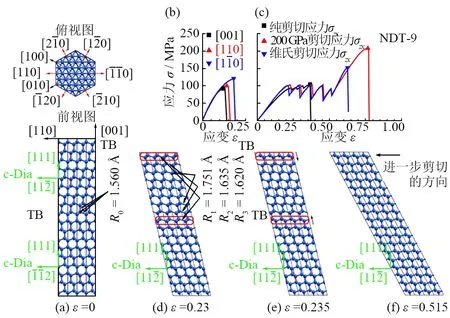
(a)纳米孪晶金刚石NTD-9模型稳定结构( 应变ε= 0 )的前视图和俯视图。红色和黑色箭头表示NTD-9中的各个晶向,而绿色箭头表示立方金刚石结构中的等效方向。(b)沿NTD-9不同拉伸方向计算获得的拉伸应力应变曲线。(c)纯剪切、非刚性维氏硬度压头以及刚性维氏硬度压头模拟计算得到的剪切应力应变曲线。(d、e、f)为非刚性维氏硬度压头沿NTD-9模型(001)[110]模拟得到的形变过程演化结构,对应的应变分别为0.230、0.235、0.515[44]。
图5不同厚度孪晶片的材料模型
图5a所示为相邻孪晶界之间包含9个C-C双层的纳米孪晶金刚石模型(NTD-9),对应的孪晶厚度为1.84 nm。对于NTD-9模型的拉伸和剪切形变的应力应变曲线如图5b和5c所示,相应形变过程的原子结构如图5d、5e、5f所示[44]。图5b拉伸应力应变曲线显示:NTD-9模型(001)面的拉伸强度最低,说明(001)面是NTD-9模型最主要的解理面。计算结果显示,(001)解理面的压痕强度与单晶金刚石(111)面的强度相当。NTD-9模型在纯剪切力作用下的峰值应力约为100 GPa,与单晶金刚石的峰值应力相当,表明没有明显的纳米孪晶增强效应。在维氏硬度压痕模型模拟过程中,若将压头设置为刚性压头得到的应力峰值达到了150 GPa,即使如此,依然远低于实验检测到的200 GPa的硬度值;若将压头设置为非刚性压头,得到的应力峰值为200 GPa,与实验结果吻合比较好。
为了讨论相应机制,LI等对刚性和非刚性维氏硬度压头模拟得到的锯齿状的应力应变曲线进行了分析。如图5d、5e、5f所示为NTD-9在非刚性维氏硬度压头沿(001)[110]方向模拟得到的一系列的结构演化过程[44]。孪晶界(TB)将模型分成上下2个部分,分别对应易剪切方向和难剪切方向。在压痕模拟中,易剪切方向上的价键拉伸得更多,而与孪晶界毗邻的R1键拉伸的最长。随着应变的增大,伸长量最多的R1键将发生翻转并重新成键,使孪晶界发生平移。如图5d和5e中的红色方框所示,价键的翻转以及重新成键使得孪晶界移动了一个原子层的位置。随着应变的增大,此过程将不断重复,孪晶界也将不断移动,直至NTD-9模型中孪晶界上部分所有的碳原子层都转到另外一边。此过程中的每一次价键的翻转和重新成键,都会使得应变能被释放,应力发生下降,并出现锯齿形的应力应变曲线,如图5c所示。一旦所有价键完成翻转并重新成键,NTD-9模型的整个结构将转变成跟单晶金刚石基本相同的结构。在此之后的压痕形变过程也将和单晶金刚石的形变过程基本相同。
为了揭示孪晶界以及孪晶片厚度对纳米孪晶金刚石硬度的影响,HUANG等[45]利用分子动力学方法模拟了纳米孪晶金刚石的纳米压痕,并研究了塑性形变的相应机制。计算发现:纳米孪晶金刚石的硬度随孪晶片厚度的减小而持续增加。在压痕下的塑性形变区域,位错在传播过程中被孪晶界阻塞以及在孪晶界一侧堆积,将导致纳米孪晶金刚石的硬化。而应变过程中,位错环沿与表面平行的方向传播,加上孪晶界的破坏而产生的更多位错成核区,都对纳米孪晶金刚石有软化效应。SUN等[46]也对纳米孪晶金刚石的力学性能和形变机制进行了分子动力学模拟。模拟得到的纳米压痕的硬度与孪晶片厚度呈正相关,但不满足在许多传统金属材料中广泛呈现的反Hall-Petch效应。目前,通过分子动力学模拟得到的结果显示:压痕下方未出现第一性原理计算得到的孪晶界迁移现象,而硬度的增强机制主要来自于孪晶界对位错的阻塞。不同小组进行的分子动力学研究结果在硬度随孪晶片厚度的变化趋势方面也有较大出入。
4 结语
纳米多晶金刚石的微观结构决定了该材料的宏观性能,而塑性形变机制的研究将揭示该材料具备优良宏观性质的内在原因,从而进一步指导设计与合成更高性能的纳米超硬材料。目前,人们利用透射电镜等技术,已经对不同实验条件下合成的纳米多晶金刚石的微观结构进行了系统的表征。但不同实验和理论研究得到的纳米多晶金刚石塑性形变机制仍存在不少争议。
实验研究方面:超高的硬度使纳米多晶金刚石很难发生塑性形变,塑性形变实验目前只能通过各种硬度压痕进行,且压痕实验中也不能使用较高的加载力,因此无法获得较大程度塑性形变的样品。超高的硬度也使得塑性形变后的纳米多晶金刚石样品后续处理存在困难。要观察塑性形变过程中的微观结构变化,目前主要的方法是在样品形变后,再对样品进行后续观测,从而推测其塑性形变的微观机制。因此,在透射电镜的制样过程中,可能会破坏样品内部本来的微观结构,人为引入其他信息,从而导致塑性形变机制的解释存在一定的偏差。
理论研究方面:纳米多晶金刚石的微观结构已经通过实验基本确定,但是在进行理论模拟时,由于考虑计算效率的因素,理论计算时的结构与材料实际情况的结构存在较大偏差,必然导致理论计算的结果与实验结果差别较大。
目前,不同课题组计算得到的纳米多晶金刚石的力学性能存在较大偏差。在纳米多晶金刚石塑性形变机制的解释方面,不同课题组提出的解释也差别较大,争论较多。因此,国内外研究人员还需不断探索,创新实验方法及检测技术,对纳米多晶金刚石的塑性形变进行更加深入的研究,揭示相应机制,促进人们对纳米超硬增强内部机制的理解,从而更好地设计和合成性能更加优异的新型超硬材料。
