吲哚类化合物的制备及抗肿瘤活性研究
2018-09-04王亚文庄方方律海峡闫福林
王亚文,庄方方,律海峡,闫福林
(1.新乡医学院药学院,河南 新乡 453003;2.河南省职工医院药学部,河南 郑州 450001)
吲哚类化合物的基本骨架为苯并吡咯环,此类化合物广泛存在于具有活性的生物体、天然产物及药物分子结构中。以吲哚为基本母核结构的化合物具有抗肿瘤[1-2]、抗炎[3]、抗菌[4]等多种药理活性。据报道,二吲哚甲烷不仅可以抑制肿瘤细胞的生长、诱导细胞的凋亡,还可抑制血管再生[5-6]。具有高效抗癌作用的药物长春新碱也具有吲哚这个基本骨架[7],这使得越来越多的药学工作者对吲哚类化合物的合成及抗肿瘤活性研究产生了浓厚兴趣。有研究报道了环β-二酮酯与吲哚间的钯催化脱氢β′功能化反应[8-10]。DMITRY等[11]将2-硝基吲哚与活泼亚甲基化合物丙二酸酯经金属锰催化,通过自由基加成反应制备出了吲哚3-位取代的化合物。随后,LOPCHUK等[12]又用2-氰基吲哚为底物,采用相似的合成路线制备出了吲哚3-位取代的产物。本课题组用硝酸铈铵催化,以乙酸乙酯为反应溶剂,以不同取代基的1,3-二羰基化合物为底物,与适量的2,2,6,6-四哌啶氧甲基化物(2,2,6,6-tetramethylpiperidinooxy,TEMPO)反应,生成α-位 TEMPO取代的β-二羰基化合物中间体,然后以价廉易得的工业原料吲哚或衍生物为底物,与α-位 TEMPO取代的β-二羰基化合物发生傅-克羟烷基化反应,制备出8种含不同取代基的吲哚三级醇化合物,并用甲基噻唑基四唑(methyl thiazolyl tetrzaolium,MTT)法对所合成的化合物进行多种肿瘤细胞株的体外抗肿瘤活性实验,现报道如下。
1 材料与方法
1.1主要试剂与仪器吲哚、乙酰乙酸乙酯和TEMPO(上海达瑞精细化学品有限公司),柱层析用乙酸乙酯、石油醚(天津科密欧试剂公司)均为分析纯,薄层层析用硅胶GF254及柱层析用硅胶(200~300目)(青岛海洋化工厂),达尔伯克改良伊格尔培养基(Dulbecco′s modified Eagle′s medium,DMEM)、胰蛋白酶培养基(美国Gibco公司),胎牛血清(杭州四季青生物技术公司),MTT、二甲基亚砜(dimethylsulfoxide,DMSO)(美国Sigma公司);Bruker-400型核磁共振波谱仪(德国Bruker公司),Q-TOF 6540高分辨质谱仪(电喷雾离子源,美国安捷伦公司),X-4显微熔点仪(温度计未校正,上海精密仪器有限公司),ZNCL-S智能恒温磁力搅拌器(河南爱博特仪器公司),ZF-7型三用紫外分析仪(上海贝仑仪器设备有限公司)。
1.2细胞株人神经母细胞瘤细胞 SHSY5Y、食管癌细胞系109、乳腺癌细胞MCF、胃癌细胞MGC均购自广州吉妮欧生物科技有限公司。
1.3吲哚类化学物合成方法
1.3.1吲哚类化学物合成路线以乙酸乙酯为反应溶剂,分别以不同取代基的1,3-二羰基化合物为底物,与适量的TEMPO反应,生成相应的α-位TEMPO取代的β-二羰基化合物中间体(图1)。然后将相应的α-位 TEMPO取代的β-二羰基化合物中间体分别与吲哚或吲哚衍生物反应,得到8个具有叔醇结构的吲哚类化合物(图2)。
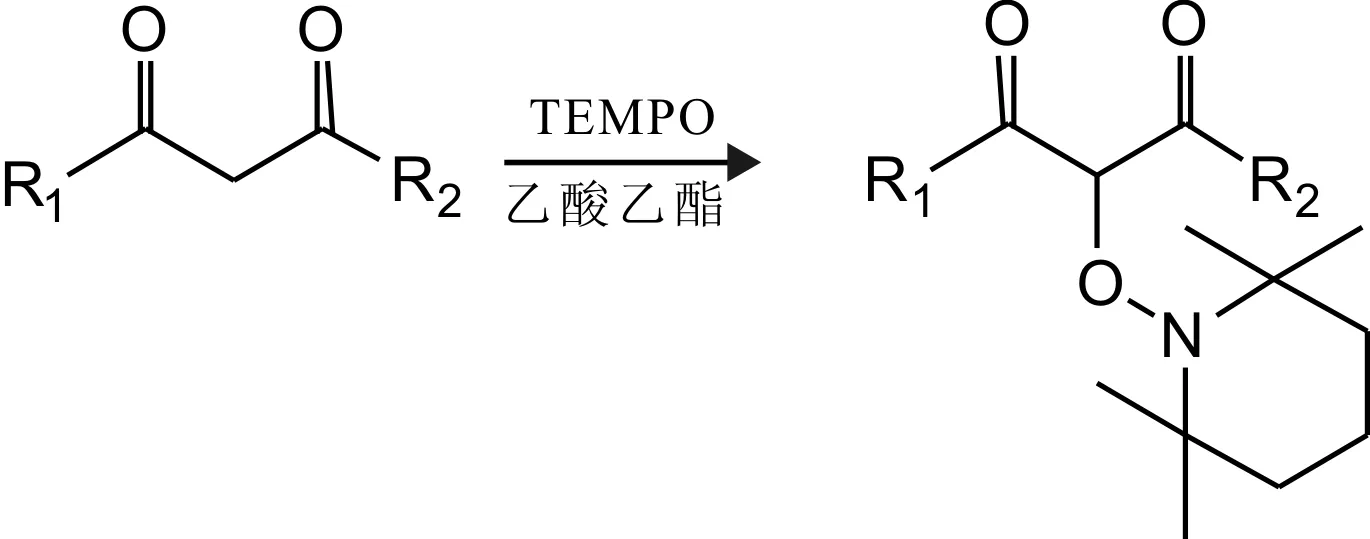
图1 α-位 TEMPO取代的β-二羰基化合物中间体的合成
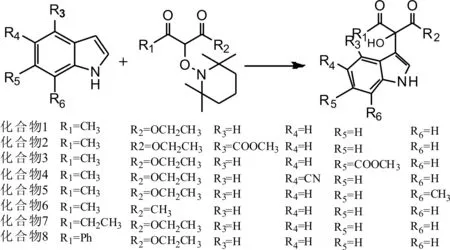
图2 吲哚类化合物的合成
1.3.2TEMPO取代的乙酰乙酸乙酯中间体的制备称取乙酰乙酸乙酯650 mg置于干燥的100 mL圆底烧瓶中,然后加入乙酸乙酯30 mL进行溶解,裸露于空气中加热,于60 ℃条件下搅拌,再称取TEMPO 780 mg缓缓加入反应瓶中,稍后加入硝酸铈铵550 mg,最后加入无水Na2SO4适量充分反应约 1.5 h,采用薄层色谱法(thin-layer chromatography,TLC)鉴定,待反应彻底后经砂芯漏斗抽滤,取其滤液经浓缩硅胶拌样,用石油醚乙酸乙酯(501)进行洗脱,通过硅胶柱层析分离纯化,得淡黄色油状产物约1 097 mg,产率85%,即TEMPO取代的乙酰乙酸乙酯中间体,核磁共振氢谱(proton nuclar magnetic resonance,1H NMR)(400 MHz,CDCl3)δ 4.79(s,1H),4.25~4.17(m,2H),2.29(s,3H),1.57~1.31(m,6H),1.26(t,J=7.1 Hz,3H),1.18(s,3H),1.17(s,3H),1.04(s,3H),0.98(s,3H)。
1.3.3化合物的制备化合物1的制备:称取制备的TEMPO取代的乙酰乙酸乙酯中间体143 mg,于50 mL圆底烧瓶中,然后加入吲哚 59 mg,用醋酸 3 mL 使其彻底溶解,将反应装置裸露于空气中置室温条件下进行搅拌,采用TLC鉴定,反应完全后加入乙酸乙酯25 mL稀释,并将反应液倒入150 mL的锥形瓶中,加入适量饱和NaHCO3进行中和,待中和完全即无气泡产生时,用乙酸乙酯(3×30 mL)进行萃取,合并有机层,加入无水Na2SO4除水,再经过滤,最后将残余液浓缩后用硅胶拌样,用石油醚乙酸乙酯(31)进行洗脱,通过硅胶柱层析分离纯化。
化合物2的制备:参照化合物1的反应条件,以4-甲酸甲酯吲哚为底物,与TEMPO取代的乙酰乙酸乙酯中间体反应。
化合物3的制备:参照化合物 1的反应条件,以6-甲酸甲酯哚为底物,与TEMPO取代的乙酰乙酸乙酯中间体反应。
化合物4的制备:参照化合物 1的反应条件,以5-氰基吲哚为底物,与TEMPO取代的乙酰乙酸乙酯中间体反应。
化合物5的制备:参照化合物1的反应条件,以7-甲基吲哚为底物,与TEMPO取代的乙酰乙酸乙酯中间体反应。
化合物6的制备:参照化合物1的反应条件,以吲哚为底物,与TEMPO取代的乙酰丙酮反应。
化合物7的制备:参照化合物1的反应条件,以吲哚为底物,与TEMPO取代的乙酰乙酸乙酯中间体反应。
化合物8的制备:称取TEMPO取代的苯甲酰乙酸乙酯化合物96 mg于50 mL圆底烧瓶中,然后加入吲哚66 mg,用3 mL醋酸使其彻底溶解,将反应装置裸露于空气中在 60 ℃ 条件下进行搅拌,经TLC鉴定,约4 h反应完全,然后加入乙酸乙酯 15 mL 稀释并将反应液倒入150 mL的锥形瓶中,加入适量饱和NaHCO3进行中和,待中和完全即无气泡产生时,用乙酸乙酯(3×30 mL)进行萃取,合并有机层,加入无水Na2SO4除水,再经过滤,最后将残余液浓缩硅胶拌样,用石油醚乙酸乙酯(31)进行洗脱,通过硅胶柱层析分离纯化。
1.4所合成化合物的抗癌活性测试
1.4.1细胞培养将人类神经母细胞瘤细胞SHSY5Y、食管癌细胞系109、乳腺癌细胞MCF、胃癌细胞MGC等细胞株用含体积分数10%胎牛血清、0.01 U·L-1链霉素和0.1 U·L-1青霉素的DMEM培养液,于37 ℃ 体积分数5%CO2培养箱中饱和湿度培养,每2~3 d换液1次,正常传代3次以后用于下一步试验。
1.4.2MMT法检测各化合物对肿瘤细胞的半数抑制浓度(halfmaximalinhibitoryconcentration,IC50) 分别取处于对数期生长、状态良好的人类神经母细胞瘤细胞SHSY5Y、食管癌细胞系109、乳腺癌细胞MCF、胃癌细胞MGC细胞,制成单细胞悬液,使细胞密度为1×102L-1,接种于96孔板,每孔加入细胞悬液100 μL,置于37 ℃ 体积分数5% CO2培养箱中培养24 h,待细胞贴壁,分为给药组、对照组、空白组。给药组按预设的浓度梯度分别加入合成的8种化合物待测样品,每个梯度6个重复。对照组加入等体积的溶解样品用的溶剂,空白组加入三蒸水,培养48 h 后,每孔加入20 μL MTT(5 g·L-1),然后置于37 ℃培养箱中温育4 h,弃上清液后加入200 μL DMSO,振荡10 min溶解沉淀,随后用酶标仪于492 nm波长处检测吸光度值,计算一定浓度下样品对细胞的抑制率。抑制率=[(对照组吸光度值-空白组吸光度值)-(给药组吸光度值-空白组吸光度值)]/(对照组吸光度值-空白组吸光度值)×100%,以抑制率为横坐标、药物浓度为纵坐标绘图,计算每个样品的IC50,IC50≤100 μmol·L-1为有肿瘤细胞抑制作用,IC50>100 μmol·L-1认为无抗肿瘤活性,数据测定无意义。实验重复3次。
2 结果
2.1化合物1分离纯化后得120 mg白色粉末状固体1,产率92%,命名为Ethyl 2-(1H-indol-3-yl)-2-hydroxy-3-oxobutanoate,白色固体,m.p.136~137 ℃。1H NMR(400 MHz,CDCl3)δ 8.39(s,1H),7.55(d,J=8.0 Hz,1H),7.37(d,J=2.5 Hz,1H),7.31(d,J=8.1 Hz,1H),7.19(t,J=7.4 Hz,1H),7.11(t,J=7.4 Hz,1H),4.81(s,1H),4.42~4.24(m,2H),2.25(s,3H),1.31(t,J=7.1 Hz,3H);核磁共振碳谱(nuclear magnetic resonance,13C NMR)(100 MHz,CDCl3)δ 204.9,170.4,136.6,125.1,124.3,122.7,120.5,120.3,111.9,111.6,82.3,62.9,25.1,14.2;C14H15NO4Na(M+Na)+高分辨质谱(high-resdution mass spectrometer,HRMS)计算值为284.089 3,实测值284.090 7。
2.2化合物2分离纯化后得到化合物2,产率67%,命名为Methyl 3-(1-ethoxy-2-hydroxy-1,3-dioxobutan-2-yl)-1H-indole-4-carboxylate,白色固体,m.p.113~114 ℃。1H NMR(400 MHz,CDCl3)δ 9.07(s,1H),7.71(d,J=7.5 Hz,1H),7.27(d,J=7.7 Hz,2H),7.07(t,J=7.8 Hz,1H),6.70(s,1H),6.37(s,1H),4.38~4.22(m,2H),3.89(s,3H),2.46(s,3H),1.31(t,J=7.1 Hz,3H);13C NMR(100 MHz,CDCl3)δ 206.3,171.1,170.6,137.9,127.6,124.1,122.3,122.3,121.2,117.4,113.4,83.6,77.4,77.1,76.8,62.4,52.7,26.2,14.0;C16H17NO4Na(M+Na)+的HRMS计算值342.094 8,实测值342.095 4。
2.3化合物3分离纯化后得到化合物3,产率90%,命名为Methyl 3-(1-ethoxy-2-hydroxy-1,3-dioxobutan-2-yl)-1H-indole-6-carboxylate,白色固体,m.p.135~136 ℃。1H NMR(400 MHz,CDCl3)δ 8.91(s,1H),8.12(s,1H),7.80(d,J=8.5 Hz,1H),7.61(d,J=8.4 Hz,2H),4.85(s,1H),4.46~4.26(m,2H),3.95(s,3H),2.25(s,3H),1.32(t,J=7.1 Hz,3H);13C NMR(100 MHz,CDCl3)δ 204.4,170.3,168.1,136.0,128.8,127.6,124.3,121.4,120.1,114.0,112.3,82.2,63.1,52.2,24.9,14.1;C16H17NO6Na(M+Na)+的HRMS计算值342.094 8,实测值342.095 1。
2.4化合物4分离纯化后得到化合物4,产率97%,命名为Ethyl 2-(5-cyano-1H-indol-3-yl)-2-hydroxy-3-oxobutanoate,灰白色固体,m.p.100~101 ℃。1H NMR(400 MHz,CDCl3)δ 9.30~8.40(br,s,1H),8.05(s,1H),7.63(s,1H),7.43(s,2H),4.82(s,1H),4.46~4.29(m,2H),2.27(s,3H),1.35(t,J=7.1 Hz,3H);13C NMR(100 MHz,CDCl3)δ 203.7,170.0,138.2,126.7,126.4,125.4,125.1,120.5,112.9,112.5,103.6,82.1,63.3,24.7,14.1;C15HN2O4Na(M+Na)+的HRMS计算值309.084 6,实测值309.086 0。
2.5化合物5分离纯化后得到化合物5,产率83%,命名为Ethyl 2-hydroxy-2-(7-methyl-1H-indol-3-yl)-3-oxobutanoate,白色固体,m.p.109~110 ℃。1H NMR(400 MHz,CDCl3)δ 8.39(s,1H),7.44~7.38(m,2H),7.11~6.96(m,2H),4.84(s,1H),4.46~4.26(m,2H),2.47(s,3H),2.27(s,3H),1.34(t,J=7.1 Hz,3H);13C NMR(100 MHz,CDCl3)δ 204.9,170.4,136.1,124.6,124.0,123.1,120.6,120.6,117.9,112.3,82.2,62.7,25.0,16.6,14.1;C15H17NO4Na(M+Na)+的HRMS计算值298.105 0,实测值298.105 4。
2.6化合物6分离纯化后得到化合物6,产率93%,命名为3-hydroxy-3-(1H-indol-3-yl)pentane-2,4-dione,白色固体,m.p.107~108 ℃。1H NMR(400 MHz,CDCl3)δ 8.39(s,1H),7.53(d,J=8.0 Hz,1H),7.38(d,J=8.2 Hz,1H),7.33(d,J=2.7 Hz,1H),7.27~7.21(m,1H),7.18~7.11(m,1H),5.36(s,1H),2.36(s,6H);13C NMR(100 MHz,CDCl3)δ 207.3,136.6,124.8,123.7,122.8,120.7,119.8,112.5,111.6,87.3,26.2;C13H13NO3Na (M+Na)+的HRMS计算值254.078 8,实测值 254.079 4。
2.7化合物7分离纯化后得到化合物7,产率93%,命名为Ethyl 2-hydroxy-2-(1H-indol-3-yl)-3-oxopentanoate,白色固体,m.p.96~97 ℃。1H NMR(400 MHz,CDCl3)δ 8.39(s,1H),7.56(d,J=8.0 Hz,1H),7.41(s,1H),7.34(d,J=8.1 Hz,1H),7.21(t,J=7.6 Hz,1H),7.12(t,J=7.5 Hz,1H),4.87(s,1H),4.45~4.24(m,2H),2.80~2.67(m,1H),2.58~2.46(m,1H),1.33(t,J=7.1 Hz,3H),1.02(t,J=7.2 Hz,3H);13C NMR(100 MHz,CDCl3)δ 207.8,170.1,136.2,124.8,124.2,122.1,119.9,119.7,111.4,111.4,81.7,62.4,30.3,13.7,7.7;C15H17NO4Na(M+Na)+的HRMS计算值298.105 0,实测值298.106 4。
2.8化合物8分离纯化后得化合物8,产率76%,命名为Ethyl 2-hydroxy-2-(1H-indol-3-yl)-3-oxo-3-phenylpropanoate,白色固体,m.p.124~125 ℃。1H NMR(400 MHz,CDCl3)δ 8.29(s,1H),8.08(d,J=8.0 Hz,2H),7.61(d,J=8.0 Hz,1H),7.50(s,1H),7.47(t,J=7.5 Hz,1H),7.32(t,J=7.1 Hz,3H),7.17(t,J=7.6 Hz,1H),7.08(t,J=7.5 Hz,1H),5.09(s,1H),4.40~4.19(m,2H),1.20(t,J=7.1 Hz,3H);13C NMR(100 MHz,CDCl3)δ 195.2,171.1,136.1,133.3,133.2,130.0,127.9,125.3,123.9,122.2,120.4,120.0,113.0,111.1,81.0,62.6,13.6;C19H17NO4Na(M+Na)+的HRMS计算值346.105 0,实测值346.105 6。
2.98种化合物体外抗肿瘤活性结果见表1。本实验合成的8种化合物中,化合物7对人神经母细胞瘤细胞SHSY5Y、食管癌细胞系109、乳腺癌细胞MCF的IC50均<100 μmol·L-1,具有较好的细胞抑制作用;化合物3和化合物6对食管癌细胞系109的IC50<100 μmol·L-1,具有一定的细胞抑制作用,对其他细胞株的细胞抑制作用较弱;化合物1、2、4、8对人神经母细胞瘤细胞SHSY5Y、食管癌细胞系109、乳腺癌细胞MCF、胃癌细胞MGC的IC50均>100 μmol·L-1,对这4种肿瘤细胞无抗肿瘤活性。化合物5对人神经母细胞瘤细胞SHSY5Y、乳腺癌细胞MCF、胃癌细胞MGC的IC50均>100 μmol·L-1,无抗肿瘤活性;对食管癌细胞系109的IC50接近100 μmol·L-1,肿瘤细胞抑制作用较弱。
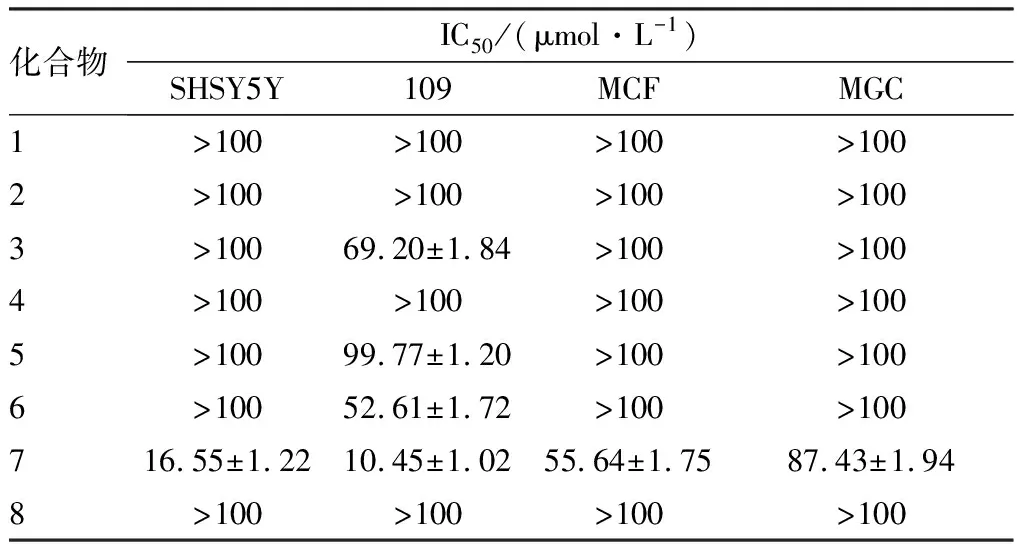
表1 化合物1~8的体外抗肿瘤活性
3 讨论
本研究探索出了一条制备含吲哚、氮杂吲哚及吡咯环的三级醇类化合物的新途径,以价廉易得的吲哚、氮杂吲哚、吡咯及制备的TEMPO取代的1,3-二羰基化合物为原料,采用傅-克羟烷基化反应,对杂环的碳-3位进行结构构建,得到8个结构新颖的杂环三级醇类化合物。该合成方法设计路线简便,反应条件温和,易于操作,收率较高,适用于大范围的各种吲哚类化合物的制备。
在对合成的8个化合物的抗肿瘤试验中发现,R1位置引入乙基和R2位置引入乙氧基的化合物7对人类神经母细胞瘤细胞SHSY5Y、食管癌细胞系109、乳腺癌细胞MCF、胃癌细胞MGC等4种细胞都有抑制活性,且对人类神经母细胞瘤细胞SHSY5Y和食管癌细胞109的抑制作用更强,化合物3和6对食管癌细胞系109具有一定的抑制作用,其余5个化合物几乎无细胞毒活性。
