基于高区别因子自组装三臂DNA纳米探针检测人体多重耐药基因
2018-09-03许榜田杨晓兰白书连
许榜田, 杨晓兰, 白书连, 赵 语*
(1.重庆医科大学附属大学城医院,重庆401331;2.重庆医科大学检验医学院,重庆400016)
癌症严重威胁着人类的健康,而多重耐药的产生是肿瘤治疗的最大障碍[1,2]。多重耐药的产生机制中最常见的是P-糖蛋白(P-gp)的过度表达。P-gp是由人类多重耐药(MDR)基因编码的一种细胞膜上的Mg2+依赖的ATP酶,其利用ATP水解释放的能量,通过细胞膜外排抗肿瘤药物。虽然大多数癌症患者可以通过化疗得到缓解,但由于耐药问题的存在,常导致一些患者化疗失败。其中多重耐药是主要原因,也是治疗白血病最大的难题[3]。此外,MDR基因也可在恶性肉瘤[4]、乳腺癌[5]患者中表达,且通常是这些患者低生存率的预警信号。研究发现MDR基因单核苷酸多态性(SNPs)可以改变P-gp蛋白构像,造成配体-受体亲和力下降。几种SNPs间可存在较强的连锁不平衡构成单倍型,导致P-gp外排功能减弱[6 - 8]。以上情况均可改变P-gp底物的代谢动力学参数[8]。此外,MDR基因SNPs还与急性淋巴细胞白血病、癌症、溃疡性结肠炎有关[9 - 11]。因此,发展准确度好、特异性高的针对MDR基因的检测方法在肿瘤早期诊断、耐药性预测和个体化治疗方面有很重要的应用前景。
传统检测MDR基因的方法有RNA印记、免疫印记、流式细胞术、荧光定量PCR等[12 - 14],尽管这些方法都有效果,但仍然存在一定的缺点,比如耗时、花费高、需要昂贵的仪器。近几年,研究者报道了许多核酸检测平台[15 - 17],设计并优化了大量的核酸检测探针[18 - 19]。Merkoçi等和Liu等报道了一种基于磁珠的荧光检测方法用于检测DNA,降低了检测成本,提高了反应速度和灵敏度[20 - 21]。Hou等报道了一种基于氮掺杂石墨烯/金纳米颗粒的DNA电化学传感器,可用于PCR样本中MDR基因的检测,并提高了灵敏度[22]。然而,这些基于核酸探针的实验最主要的缺陷是缺乏特异性和通用性。Kool等报道了荧光模板化的级联反应用于RNA的检测,虽然提高了检测的灵敏度并具有一定的特异性,但需要复杂的核酸预处理和化学连接[23]。Tyagi等人提出的分子信标是一种由一条单链DNA组成的可在常温条件下形成颈环二级结构的检测探针[24],与单链探针相比,分子信标虽具有一定特异性,但其设计困难且无法检测基因组长片段DNA。因此,迫切需要建立一种新颖的方法实现特异、准确并快速地检测MDR基因。
近来,文献报道双链探针可作为核酸错配特异识别探针与目标物发生基于Toehold交换的链替代反应[25 - 26]。在合适的条件下,双链探针与目标物自发地杂交,且由单碱基错配产生的微小的热力学参数改变即可对链替代效率产生极大影响,从而实现高特异性检测目标物。本研究构建了一种高特异性且通用的三臂DNA荧光探针。设计的探针在检测化学合成MDR基因片断时展现出良好的特异性。本实验不需要酶的辅助且反应条件温和,易于在临床推广。此外,该方法对PCR产物及体液中单链DNA或RNA的特异、精准检测有着广阔的应用前景。
1 实验原理
基于三臂DNA纳米结构的MDR基因检测原理示意图如图1所示,目标识别部分和信号报告部分由两个相互独立的杂交反应组成,并形成一种稳定的三臂DNA纳米结构。该结构中修饰猝灭基团的S1与修饰荧光基团的S2相互靠近,使荧光信号猝灭。当体系中存在MDR基因时,其与Toehold稳定结合进而发生链替代反应将保护链从互补链上置换下来,使S1和S2分离并产生荧光信号。当MDR基因链中与Toehold互补的区域发生突变时,MDR基因与检测探针结合不稳定,难以引发链替代反应。

图1 基于三臂DNA纳米结构的MDR基因检测原理示意图Fig.1 Working principle of three-arm DNA nanoprobe for MDR gene detection
2 实验部分
2.1 仪器与试剂
Agilent Cary Eclipse荧光分光光度计(美国);DYY-6C电泳仪(北京市六一仪器厂);Bio-Rad Laboratories凝胶成像系统(美国)。
过硫酸铵(AP)及四甲基乙二胺(TEMED)(美国Sigma公司);核酸染料(日本Takara公司);所有核酸均购自上海生工生物工程有限公司,其序列见表1;其他试剂均为分析纯。实验用水均为去离子水。
2.2 信号检测
将保护链(P1)、互补链(C1)、荧光猝灭链(S1)和荧光链(S2)混合,并用杂交液补充至150 μL,在90 ℃孵育5 min后,立即放在冰上。用不同的处理条件处理,加入50 μL不同浓度的样本在对应条件下孵育,在激发和发射狭缝均为10 nm、激发波长为496 nm、发射波长为520 nm的条件下测定上述溶液的荧光强度。所有实验均重复测定3次。
2.3 非变性聚丙烯酰胺凝胶电泳(Native Polyacrylamide Gel Electrophoresis,native-PAGE)
配制8%的非变性聚丙烯酰胺凝胶,在37 ℃浮箱中孵育30 min后取出,每孔按5∶1的比例加入终浓度为1.0 μmol/L的无修饰核酸杂交液和上样缓冲溶液,在90 mV电压条件下进行65 min后取出胶条,加入染色液避光染色30 min。用凝胶成像系统曝光。

表1 DNA或RNA序列
The bold bases are mismatched bases or delete bases.
2.4 荧光动力学实验
150 μL荧光标记的探针与50 μL不同浓度的样本或50 μL缓冲溶液在石英比色皿中混合均匀后,立即在激发和发射狭缝均为10 nm、激发波长为496 nm、发射波长为520 nm的条件下测定上述溶液的荧光强度。所有实验均重复测定3次。
3 结果与讨论
3.1 三臂DNA纳米结构的表征与实验可行性分析
3.1.1三臂DNA纳米结构的荧光表征与可行性分析首先,用荧光动力学实验表征三臂DNA纳米结构并分析实验可行性(图2(A))。当加入FAM-S2后,荧光分光光度计检测到明显的荧光信号。图2(A)曲线b表明,向其中加入缓冲液后,荧光信号因稀释效应而下降。当其中加入等体积Dabcyl-S1/P1杂交产物时,曲线a与曲线b荧光信号差别不大。当其中单独加入等体积Dabcyl-S1时,曲线c荧光信号下降较曲线a、b明显,表明除稀释作用外,游离的Dabcyl-S1与游离的FAM-S2之间可能有部分非特异杂交产物,使荧光下降。当其中加入等体积Dabcyl-S1/P1/C1杂交产物时,荧光信号下降非常明显,表明在溶液中形成了三臂DNA纳米结构(曲线d、e),使S2上的荧光基团FAM与S1上的猝灭基团Dabcyl相互靠近,荧光被猝灭。当再次加入缓冲液后,荧光信号因稀释作用而下降(曲线d)。而加入相等体积的MDR基因后,荧光信号先因稀释作用而下降,后又因MDR基因与三臂DNA纳米结构之间发生反应,三臂DNA纳米结构裂解使S2上的荧光基团FAM与S1上的猝灭基团Dabcyl分离,荧光信号恢复。
核酸杂交的热力学参数如表2所示。

表2 热力学参数
3.1.2三臂DNA纳米结构的native-PAGE表征如图2(B)所示,三臂DNA纳米结构的形成同样被native-PAGE证明。为避免荧光染料对电泳过程的干扰,此实验S1和S2均未修饰猝灭基团和荧光基团。泳道6上的条带(P1/C1)迁移速度低于泳道2条带(P1)迁移速度,表明P1/C1杂交产物形成。泳道7上的条带(P1/C1/S1)迁移速度低于泳道6条带迁移速度,表明P1/C1/S1杂交产物形成。泳道8上的条带(P1/C1/S1/S2)迁移速度低于泳道7上的条带迁移速度,表明三臂DNA纳米结构的形成。

图2 (A)三臂DNA纳米探针核酸链替代反应的荧光动力学表征;(B)三臂DNA纳米探针的非变性聚丙烯酰氨凝胶电泳表征Fig.2 (A)Characterization of toehold-mediated strand displacement reaction by dynamic fluorescence detection;(B)Nucleic acid hybridization reaction by native-PAGE(A)a.S2(100 μL,200 nmol/L)+S1(50 μL,400 nmol/L)+P1(50 μL,400 nmol/L);b.S2(100 μL,200 nmol/L)+buffer(50 μL);c.S2(100 μL,200 nmol/L)+S1(50 μL,400 nmol/L);d.S2(100 μL,200 nmol/L)+S1(50 μL,400 nmol/L)+P1(50 μL,400 nmol/L)+C1(50 μL,400 nmol/L)+buffer(50 μL);e.S2(100 μL,200 nmol/L)+S1(50 μL,400 nmol/L)+P 1(50 μL,400 nmol/L)+C1(50 μL,400 nmol/L)+MDR gene(50 μL,400 nmol/L);(B)1.20 bp DNA ladder;2.P1;3.C1;4.S1;5.S2;6.P1+C1;7.P1+C1+S1;8.P1+C1+S1+S2;the final concentration of nucleic acid is 1.0 μmol/L.
3.2 实验条件的优化
如图3所示,对反应体系中的核酸链P1与C1浓度的比例、S1与S2浓度的比例、孵育时间、孵育温度进行了优化。核酸链P1与C1的比例对信噪比的影响见图3(A),P1与C1浓度的最优比为0.8∶1。超过或低于这一比例时,信噪比逐渐降低。因而S1与S2最优比选择0.8∶1。S1与S2的比例对信噪比的影响见图3(B),S1与S2浓度的最优比为2∶1。超过或低于这一比例时,信噪比逐渐降低。因而S1与S2最优比选择2∶1。图3(C)对孵育时间进行了优化,当加入相同浓度和体积的MDR基因后,37 ℃条件下,孵育时间在30 min左右时荧光强度达到平衡。因此后续试验选择30 min孵育时间。孵育温度对荧光信号的影响如图3(D)所示, 37 ℃时荧光强度最强,因此后续试验选择37 ℃作为孵育温度。

图3 (A)P1与C1浓度比对信噪比的影响;(B)S1与S2浓度比对信噪比的影响;(C)孵育时间对荧光强度的影响;(D)孵育温度对荧光强度的影响Fig.3 (A)Signal-to-noise ratio at various concentration ratio of P1 and C1;(B)Signal-to-noise ratio at various concentration ratio of S1 and S2;(C)Fluorescence intensity at different incubation time;(D)Fluorescence intensity at different incubation temperatures(A)S1,200 nmol/L;S2,200 nmol/L;MDR,50 nmol/L;(B)P1,160 nmol/L;C,200 nmol/L;MDR,50 nmol/L;(C)S1,400 nmol/L;S2,200 nmol/L;P1,160 nmol/L;C1,200 nmol/L;MDR,50 nmol/L;(D)S1,400 nmol/L;S2,200 nmol/L;P1,160 nmol/L;C1,200 nmol/L.MDR,50 nmol/L.All the experiments were repeated thrice.
3.3 三臂DNA纳米结构检测性能分析
如图4所示,本实验能够明显区分野生型MDR基因与突变型MDR基因。为验证本方法对MDR基因点突变的特异识别能力,用区别因子(DF),DF=ΔFwild/ΔFmutation(Δ代表除去背景信号)对野生型MDR基因及2m,3m,3d,5m,5d几种突变型MDR基因进行评估。在浓度相同的情况下,三臂DNA纳米探针的最高区别因子为24.1,最低区别因子为4.1,平均为11.8。更重要的是,其对不同突变位点的MDR基因展示出明显的区别因子,且对同一位点的不同突变类型也展示出良好的区别因子。该方法最高特异性区别能力与其他检测方法相比有明显提高[27]。以上结果说明本实验有很好的单碱基突变识别能力。尽管许多方法被用于提高核酸检测的特异性,例如分子信标[28]、DNA酶的特异性识别[29]、化学修饰[30]以及级联链替代反应[31]等,但是以上方法或不适合检测长片段目标DNA,或价格昂贵不适合临床推广。

图4 (A,B)三臂DNA纳米探针检测野生型MDR基因和不同位点突变型MDR基因的荧光强度;(C)三臂DNA纳米探针对野生型MDR基因和不同位点突变型MDR基因的区别能力Fig.4 (A,B)Fluorescence intensity of wild MDR gene and mutant MDR gene at different mutational site;(C)The specificity of probe for MDR mutation genes was evaluated by DFs(A,B)S1,400 nmol/L;S2,200 nmol/L;P,160 nmol/L;C,200 nmol/L;wild MDR gene and mutant MDR gene,50 nmol/L.All the experiments were repeated thrice.
如图5(A)所示,在最佳条件下,用动力学方法检测不同浓度MDR基因对荧光信号的影响,荧光强度与MDR基因浓度有很好的线性关系。如图5(B)所示,用终点法检测不同浓度的MDR基因,获得标准曲线。其线性方程为:y=2.0709x+1.1963(R2=0.9825)。其中,y为荧光强度,x为MDR基因浓度(nmol/L)。说明荧光强度随着MDR基因浓度的增加而增加。

图5 (A)检测方法对不同浓度MDR基因的相应曲线;(B)检测方法的校正曲线Fig.5 (A)Fluorescence curves of this method at different concentration of MDR gene;(B)The calibration curve of this methodfrom the bottom to up:0 nmol/L to 120 nmol/L;S1,400 nmol/L;S2,200 nmol/L;P,160 nmol/L;C,200 nmol/L.All the experiments were repeated thrice.
3.4 三臂DNA纳米结构的通用性实验
为了证明本方法的通用性,对miR-21进行了检测。如图6(A)所示,先用UNA fold software(http://mfold.rna.albany.edu/)模拟了S2 与 C′,S1 与 P′ 之间的杂交,显示无其他二级结构产生。如图6(B)所示,用该探针对miR-21进行检测,野生型miR-21产生很强的荧光信号,而突变型miR-21的荧光信号与野生型miR-21相比明显降低,几乎与空白对照组荧光信号相同,表明突变型miR-21不能有效的与三臂DNA纳米结构发生链替代反应。这一实验表明,三臂DNA纳米探针在不需要重新设计信号报告部分的前提下,可用于检测miR-21。同时也说明本方法具备检测其他目标DNA的能力,有很好的通用性。

图6 (A)软件模拟S2与C′,S1与P′杂交产物;(B)检测野生型与突变型miR-21Fig.6 (A)Hybridization product between S2 and C′,S1 and P′ evaluated by software;(B)Wild type and mutant miR-21 detectionS1:400 nmol/L;S2:200 nmol/L;P′:160 nmol/L;C′:200 nmol/L;miR-21 and 3d′:100 nmol/L.All the experiments were repeated thrice.
3.5 样本回收率测定
采用标准加入法对本方法进行回收率测定。从重庆医科大学附属大学城医院大公馆分院检验科收集人血清,加入3个不同浓度梯度的化学合成MDR基因样本,按本方法进行测定,重复3次测定的平均回收率为99.0%~101.7%(表3),说明此方法具备对实际样本的检测能力。
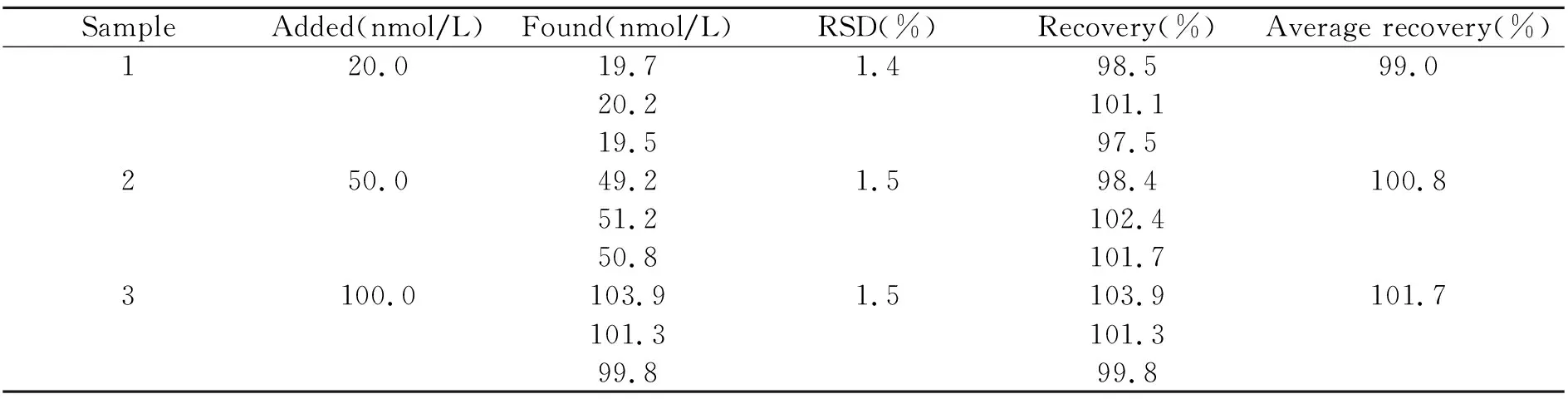
表3 MDR基因回收率测定(n=3)
4 结论
本研究构建了一种新型的三臂DNA纳米探针检测MDR基因,其最高区别因子为24.1,最低区别因子为4.1,平均区别因子为11.8。对不同突变位点的MDR基因和对同一位点不同突变类型的MDR基因,三臂DNA纳米探针均展示出明显的区别因子。用标准加入法检测混合人血清中MDR基因时,回收率在99.0%~101.7%之间。此外,本实验不需重新设计信号报告链即可实现对miR-21的检测。本方法特异性高、通用性好、不需酶的辅助且条件温和,为单链PCR产物及体液中单链DNA或RNA的精准检测提供了有力的技术支撑。
